Maybe you’ve seen the movies or played with toy Transformers, those shape-shifting machines that morph in response to whatever challenge they face.It turns out that DNA-repair machines in your cells use a similar approach to fight cancer and other diseases, according to research led by scientists from the U.S. Department of Energy’s Lawrence Berkeley National Laboratory (Berkeley Lab).
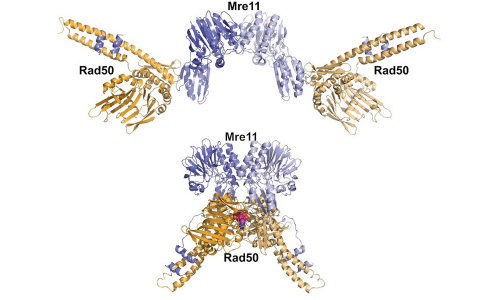
As reported in a pair of new studies, the scientists gained new insights into how a protein complex called Mre11-Rad50 reshapes itself to take on different DNA-repair tasks.
Their research sheds light on how this molecular restructuring leads to different outcomes in a cell. It could also guide the development of better cancer-fighting therapies and more effective gene therapies.
Mre11-Rad50’s job is the same in your cells, your pet’s cells, or any organism’s. It detects and helps fix the gravest kind of DNA breaks in which both strands of a DNA double helix are cut. The protein complex binds to the broken DNA ends, sends out a signal that stops the cell from dividing, and uses its shape-shifting ability to choose which DNA repair process is launched to fix the broken DNA. If unrepaired, double strand breaks are lethal to the cell. In addition, a repair job gone wrong can lead to the proliferation of cancer cells.
Little is known about how the protein’s Transformer-like capabilities relate to its DNA-repair functions, however.
To learn more, the scientists modified the protein complex in ways that were designed to affect just one of the many activities it undertakes. They then used structural biology, biochemistry, and genomic tools to study the impacts of these modifications.
“By targeting a single activity, we can make the protein complex go down a different pathway and learn how its dynamic structure changes,” says John Tainer of Berkeley Lab’s Life Sciences Division. He conducted the research with fellow Berkeley Lab scientist Gareth Williams and scientists from several other institutions.
Adds Williams, “In some cases, we sped up or slowed down the protein complex’s movements, and by doing so we changed its biological outcomes.”
Much of the research was conducted at the Advanced Light Source (ALS), a synchrotron located at Berkeley Lab that generates intense X-rays to probe the fundamental properties of substances. They used an ALS beamline called SYBILS, which combines X-ray scattering with X-ray diffraction capabilities. It yields atomic-resolution images of the crystal structures of proteins. It can also watch the transformation of the protein as it undergoes conformational changes.
In one study published in the journal Molecular Cell, the scientists studied Mre11 from microbial cells. They developed two molecular inhibitors that block Mre11’s ability to cut DNA, a critical initial step in the repair process.
They tested the effect of these inhibitors in human cells. They found that Mre11 first makes a nick away from the broken DNA strand it is repairing. Mre11 then works back toward the broken end. Previously, scientists thought that Mre11 always starts at the broken DNA end. They also found that when Mre11 cuts in the middle of a DNA strand, it initiates a high-precision DNA-repair pathway called homologous recombination repair.

In another study published in EMBO Journal, the scientists created Rad50 mutations that either promote or destabilize the shape formed when the Rad50 subunit binds with ATP, a chemical that fuels the protein complex’s movements.
Biochemical and functional assays conducted by Tanya Paull of the University of Texas at Austin revealed how these changes affect microbial, yeast, and human Mre11-Rad50 activities. Paul Russell at the Scripps Research Institute helped the scientists learn how these Rad50 mutations affect yeast cells.
They found that some mutations slowed down ATP hydrolysis, which is how Rad50 and other enzymes use ATP as fuel. Other mutations sped it up. Both changes affected Mre11-Rad50’s workflow, and its biological outcomes, in a big way.
“When we slowed down hydrolysis and favored the ATP-bound state, Rad50 favoured a non-homologous end joining pathway, which is a low-fidelity way to repair DNA,” says Williams. “When we sped it up, the subunit favoured homologous repair, which is the high-fidelity pathway.”
This approach, in which scientists start with a specific protein mechanism and learn how it affects the entire organism, will help researchers develop a predictive understanding of how Mre11-Rad50 works.
“It’s a ‘bottom up’ way to study proteins such as Mre11-Rad50, and it could guide the development of better cancer therapies and other applications,” says Tainer.
Story Source:
The above story is based on materials provided by U.S. Department of Energy’s Lawrence Berkeley National Laboratory.
{kind=link}