A computational method for finding transition states in chemical reactions, greatly reducing computational costs with high reliability, has been devised. Compared to the most widely used existing method, the present method reduces the total computational cost by approximately 50 to 70%. The development, available on GitHub, is poised to accelerate advancements in material science, making the exploration of chemical reactions more accessible and efficient. This could lead to faster scientific discoveries and technological innovations.
Credit: Shin-ichi Koda
A computational method for finding transition states in chemical reactions, greatly reducing computational costs with high reliability, has been devised. Compared to the most widely used existing method, the present method reduces the total computational cost by approximately 50 to 70%. The development, available on GitHub, is poised to accelerate advancements in material science, making the exploration of chemical reactions more accessible and efficient. This could lead to faster scientific discoveries and technological innovations.
In chemical reactions, substances transform from one energetically stable state to another, passing through an unstable transition state. This process is akin to finding the lowest elevation route over a mountain when crossing from one side to the other. Understanding the transition state – the peak of this metaphorical mountain path – is crucial for a profound comprehension of reaction mechanisms. However, due to the transient and unstable nature of these states, their experimental observation and identification are challenging, often necessitating computational exploration.
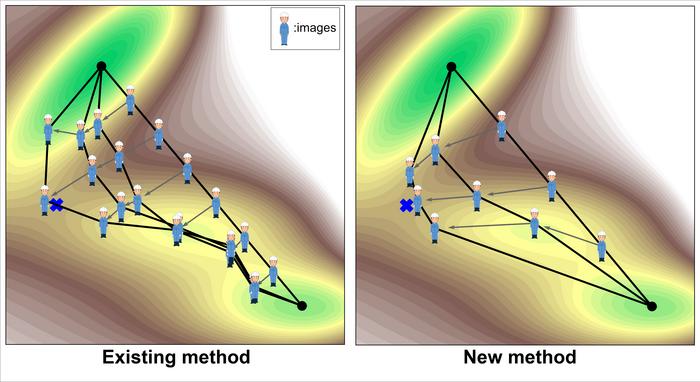
This study focuses on computational methods for finding a transition state between a known reactant and product. This type of transition state search optimizes the path connecting the product and reactant so that it passes through the transition state. Since the path is usually represented by multiple points on the path (often called images, metaphorically represented by people in Figure 1), the path is actually optimized by incrementally updating the images.
The most commonly used method today is the Nudged Elastic Band (NEB) method (Figure 1, left). One of the main challenges of this method is that it is computationally expensive. There are two main reasons for this. One is that it requires a large number of images to increase the resolution of the search. The other reason is that the search principle is not variational (i.e., minimizing an objective function), so the number of updates per image also tends to be large.
The newly implemented method in this study innovatively solves these problems (Figure 1, right). First, the number of images can be reduced to about 3, since only the region around the transition state is intensively searched. In addition, the search principle is variational, so it can be solved more efficiently. Specifically, the objective function is defined as the line integral of the exponential of the energy along the path.
The performance of our new method was evaluated on 121 chemical reactions and the results were compared with the NEB method and its improved version. First, the present method correctly identified transition states in 98% of the cases. This accuracy is much higher than the NEB method and comparable to the improved version. Second, the present method showed a significant reduction in total computational cost – about 70% less than the NEB method and 50% less than its improved version.
To facilitate wider application, we have made our computational program available on GitHub (github.com/shin1koda/dmf). Written in Python and designed to be used with the Atomic Simulation Environment (ASE), it allows researchers to easily explore transition states by specifying reactants and products.
Looking ahead, the implications of this research are vast. By making transition state searches easier and faster, our method is poised to accelerate researches and developments in all fields of natural science using computational chemistry.
Information of the paper:
Authors: Shin-ichi Koda and Shinji Saito
Journal Name: Journal of Chemical Theory and Computation
Journal Title: “Locating Transition States by Variational Reaction Path Optimization with an Energy-Derivative-Free Objective Function”
DOI: 10.1021/acs.jctc.3c01246
Journal
Journal of Chemical Theory and Computation
DOI
10.1021/acs.jctc.3c01246
Method of Research
Data/statistical analysis
Subject of Research
Not applicable
Article Title
Locating Transition States by Variational Reaction Path Optimization with an Energy-Derivative-Free Objective Function
Article Publication Date
22-Mar-2024


{kind=link}