LA JOLLA (February 7, 2024)—Lung adenocarcinoma is the most common lung cancer and the cause of most cancer-related deaths in the United States. There are several ways lung adenocarcinoma can arise, one of which is a mutation in a protein called EGFR (epidermal growth factor receptor). Non-mutated EGFR helps cells grow in response to injury, but mutated EGFR promotes out-of-control growth that can turn into cancer. Modern immunotherapies don’t work against EGFR-driven lung adenocarcinoma, and while some drugs exist to treat the cancer, patients typically develop a resistance to them within just a few years. This gap in the treatment toolchest inspired Salk Institute researchers to probe for weak spots in the cancer’s growth pathway.
Credit: Salk Institute
LA JOLLA (February 7, 2024)—Lung adenocarcinoma is the most common lung cancer and the cause of most cancer-related deaths in the United States. There are several ways lung adenocarcinoma can arise, one of which is a mutation in a protein called EGFR (epidermal growth factor receptor). Non-mutated EGFR helps cells grow in response to injury, but mutated EGFR promotes out-of-control growth that can turn into cancer. Modern immunotherapies don’t work against EGFR-driven lung adenocarcinoma, and while some drugs exist to treat the cancer, patients typically develop a resistance to them within just a few years. This gap in the treatment toolchest inspired Salk Institute researchers to probe for weak spots in the cancer’s growth pathway.
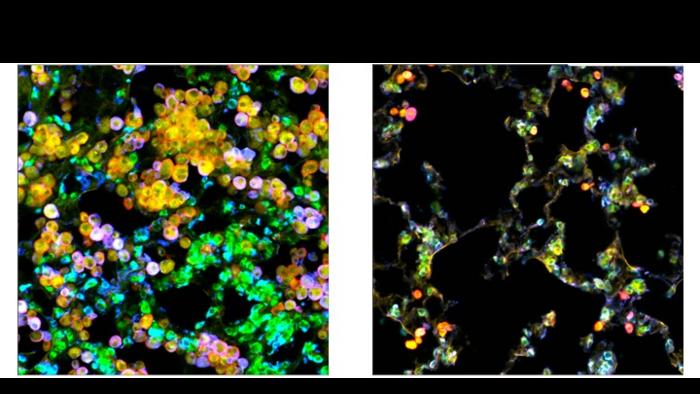
The team discovered that EGFR-driven lung adenocarcinoma hijacks a specialized population of lung-resident immune cells called macrophages, which are designed to dispose of diseased and damaged cells, as well as maintain a delicate balance of protective lipids (fats) around lung alveoli, which are essential for breathing. The lung cancer cells pull macrophages into the tumor microenvironment and alter their lipid metabolism to turn them into cancer fuel-suppliers. The newly energized tumor cells then spur further macrophage proliferation to supply more fuel—a novel self-perpetuating cancer mechanism.
The findings, published in Cancer Discovery on January 25, 2024, provide new inspiration for lung adenocarcinoma interventions that disrupt this tumor cell-macrophage relationship. The researchers suggest that treatments using EGFR inhibitors may be more successful when paired with statins, a class of drugs commonly used to lower cholesterol levels.
“We have discovered a novel way that lung cancer cells manipulate their local environment and other cell types surrounding them to promote their own growth. In this case, the tumor cells exploit lung-resident macrophages—remodeling them to provide nutrients, like cholesterol, to the cancer cells and stimulate tumor growth,” says senior and co-corresponding author Susan Kaech, professor, director of the NOMIS Center for Immunobiology and Microbial Pathogenesis, and holder of the NOMIS Chair at Salk. “One exciting implication of this work is that lung cancer treatments may be improved by simply adding statins, an already widely used class of drugs, to the patient’s treatment plan.”
Lungs rely on tiny bulbs called alveoli, which expand and deflate with our breath, to facilitate the exchange of oxygen and carbon dioxide between the air and our blood. Alveoli are crucial to human survival, and their health is dependent on a lipid-rich environment created by alveolar cells and sustained by macrophages. Lipids, such as cholesterol, are fatty compounds that support bodily function by helping cells move, store energy, and absorb vitamins.
This unique ability of lung-resident macrophages to maintain lipid balance becomes more complicated when tumor cells begin to exploit those lipids to help themselves grow. A better understanding of the mechanisms macrophages use to regulate their metabolism and lipid production can provide insight into how tumor cells selfishly manipulate those mechanisms to help themselves.
“The tumor cells excrete even more of a growth factor called GM-CSF (granulocyte macrophage colony-stimulating factor), which then causes the macrophages to grow alongside them and change their metabolism, resulting in excess lipids that the tumor cells use to strengthen themselves,” says first author Alexandra Kuhlmann-Hogan, former postdoctoral researcher in Kaech’s lab and current postdoctoral researcher at UC Los Angeles. “The cancer was effectively hijacking this normal macrophage process of maintaining the lungs with healthy lipids in order to fuel itself.”
“Not only were the tumor cells metabolically reprogramming the macrophages—they were also instigating a feedback loop that encouraged an optimal metabolic state in the tumor cells themselves,” says co-corresponding author Katerina Politi, scientific director of the Center for Thoracic Cancers at Yale Cancer Center and professor of pathology at Yale School of Medicine.
When the EGFR-driven lung adenocarcinoma cells secreted GM-CSF, it stimulated a gene in the macrophages called PPARγ (peroxisome proliferator-activated receptor gamma), which jump-started their metabolic reprogramming and subsequent secretion of lipids. In addition to using these macrophage-curated lipids to grow, the tumor cells also use the lipids to power the continued activation of the EGFR-drive that helps the cancer grow.
Kaech predicts that disrupting this loop could be a novel intervention for slowing down EGFR-driven cancer growth. Exactly how the delivery of lipids like cholesterol to tumor cells powers the EGFR oncogenic pathway, the researchers aren’t yet sure.
“Our results reveal new therapeutic possibilities for immunotherapy-resistant EGFR-driven lung adenocarcinomas,” says co-corresponding author Christian Metallo, professor and holder of the Daniel and Martina Lewis Chair at Salk. “We have identified a key metabolic relationship between macrophages and alveoli that is exploited by tumor cells to support the cancer’s metabolic demands—now we just have to disrupt that exploitation.”
In future clinical trials, the researchers recommend pairing PPARy inhibitors, which would disrupt macrophage hijacking, with statins, which would limit available cholesterol along with the currently used EGFR inhibitors. They are also curious whether similar immunological hijacking occurs in other tumor microenvironments around the body, suggesting these results may prompt further discoveries across other cancer types and immune cells.
Other authors include Ziyan Xu, Ramya Kuna, Kacie Traina, Anna-Maria Globig, and Reuben Shaw of Salk; Thekla Cordes of Technishe Universität Braunschweig in Germany; Elizabeth Kwong and Sandra Leibel of UC San Diego School of Medicine and Sanford Consortium for Regenerative Medicine; Matthew Nobari and George Cheng of UC San Diego Department of Medicine; and Camila Robles-Oteíza, Deborah Ayeni, Stellar Levy, and Robert Homer of Yale School of Medicine.
The work was supported by the National Institutes of Health (RO1CA230275, RO1CA195720, R35 R35CA220538, R01CA234245, RO1 CA216101S1), the Yale Cancer Biology Training Program (T32 CA193200-01A1), Mark Foundation for Cancer Research, Yale University Interdisciplinary Immunology Training Grant (T32 AI-007019), NOMIS Foundation, Waitt Foundation, Chapman Foundation, Helmsley Charitable Trust, and Helmsley Center for Genomic Medicine.
About the Salk Institute for Biological Studies:
Unlocking the secrets of life itself is the driving force behind the Salk Institute. Our team of world-class, award-winning scientists pushes the boundaries of knowledge in areas such as neuroscience, cancer research, aging, immunobiology, plant biology, computational biology, and more. Founded by Jonas Salk, developer of the first safe and effective polio vaccine, the Institute is an independent, nonprofit research organization and architectural landmark: small by choice, intimate by nature, and fearless in the face of any challenge. Learn more at www.salk.edu.
Journal
Cancer Discovery
DOI
10.1158/2159-8290.CD-23-0434
Article Title
EGFR-driven lung adenocarcinomas coopt alveolar macrophage metabolism and function to support EGFR signaling and growth
Article Publication Date
25-Jan-2024

