The photocatalytic conversion of methane with a ubiquitous and clean oxidant of water has the potential to develop into an on-site and on-demand chemical technology for the green utilization of methane in an environmentally benign and sustainable way. However, rational design of next-generation photocatalysts is hindered by the lack of molecular-level understanding of hole-driven oxidation kinetics, active sites, and resultant photocatalytic performance.
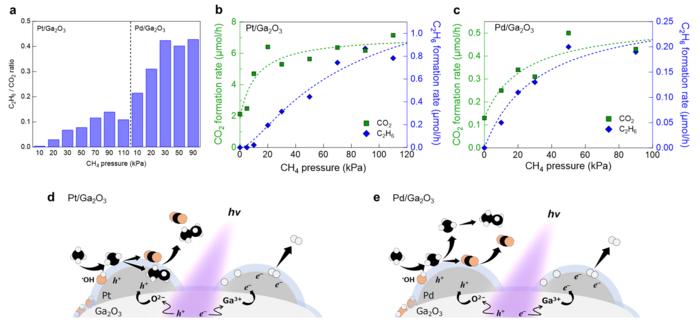
The research group led by Toshiki Sugimoto, Associate Professor at the Institute for Molecular Science, has demonstrated that metal cocatalysts loaded on a semiconductor photocatalyst play critical roles in modulating surface oxidation kinetics and resultant oxidation selectivity. Real-time mass spectrometric analysis of gaseous products under systematically-controlled methane pressures revealed that the Pt-loaded Ga2O3 photocatalyst predominantly promoted the total oxidation of methane toward CO2 on its surface, while the Pd-loaded photocatalyst exhibited a higher selectivity for C2H6 formation through the gas-phase coupling of free •CH3 (Figure 1). This difference in methane oxidation kinetics was corroborated by operando infrared absorption spectroscopy that observed surface intermediates under working conditions (Figure 2). Moreover, the research group demonstrated that the Pt cocatalyst itself was oxidized by photogenerated holes. These experimental results demonstrated the critical roles of metal cocatalysts as a reservoir of photogenerated holes and an effective reaction site for methane oxidation processes.
Generally, metal cocatalysts have been recognized for half a century as reduction cocatalysts that exclusively accumulate photogenerated electrons and promote reduction reactions such as H2 evolution. Based on this conventional assumption, hole-accumulated metal cocatalysts are assumed to act as charge recombination centers and inhibit photocatalysis. In contrast, this research group verified that both H2 evolution and methane oxidation were accelerated by metal cocatalyst loading. These experimental results indicate that photogenerated electrons and holes are separately trapped at different metal cocatalyst particles while avoiding charge recombination and promoting redox reactions (Figure 1d, e).
Thus, the systematic operando investigation of the photocatalytic oxidation of methane (i.e., the most inert and simplest organic compound) and water (i.e., one of the key molecules in photocatalysis) provides a new paradigm for the role of metal cocatalysts in photocatalysis and thus contributes to developing a cocatalyst-based surface engineering strategy for controlling non-thermal oxidation reactions.
Credit: Toshiki Sugimoto
The photocatalytic conversion of methane with a ubiquitous and clean oxidant of water has the potential to develop into an on-site and on-demand chemical technology for the green utilization of methane in an environmentally benign and sustainable way. However, rational design of next-generation photocatalysts is hindered by the lack of molecular-level understanding of hole-driven oxidation kinetics, active sites, and resultant photocatalytic performance.
The research group led by Toshiki Sugimoto, Associate Professor at the Institute for Molecular Science, has demonstrated that metal cocatalysts loaded on a semiconductor photocatalyst play critical roles in modulating surface oxidation kinetics and resultant oxidation selectivity. Real-time mass spectrometric analysis of gaseous products under systematically-controlled methane pressures revealed that the Pt-loaded Ga2O3 photocatalyst predominantly promoted the total oxidation of methane toward CO2 on its surface, while the Pd-loaded photocatalyst exhibited a higher selectivity for C2H6 formation through the gas-phase coupling of free •CH3 (Figure 1). This difference in methane oxidation kinetics was corroborated by operando infrared absorption spectroscopy that observed surface intermediates under working conditions (Figure 2). Moreover, the research group demonstrated that the Pt cocatalyst itself was oxidized by photogenerated holes. These experimental results demonstrated the critical roles of metal cocatalysts as a reservoir of photogenerated holes and an effective reaction site for methane oxidation processes.
Generally, metal cocatalysts have been recognized for half a century as reduction cocatalysts that exclusively accumulate photogenerated electrons and promote reduction reactions such as H2 evolution. Based on this conventional assumption, hole-accumulated metal cocatalysts are assumed to act as charge recombination centers and inhibit photocatalysis. In contrast, this research group verified that both H2 evolution and methane oxidation were accelerated by metal cocatalyst loading. These experimental results indicate that photogenerated electrons and holes are separately trapped at different metal cocatalyst particles while avoiding charge recombination and promoting redox reactions (Figure 1d, e).
Thus, the systematic operando investigation of the photocatalytic oxidation of methane (i.e., the most inert and simplest organic compound) and water (i.e., one of the key molecules in photocatalysis) provides a new paradigm for the role of metal cocatalysts in photocatalysis and thus contributes to developing a cocatalyst-based surface engineering strategy for controlling non-thermal oxidation reactions.
Journal
Angewandte Chemie International Edition
DOI
10.1002/anie.202306058
Method of Research
Experimental study
Subject of Research
Not applicable
Article Title
Beyond Reduction Cocatalysts: Critical Role of Metal Cocatalysts in Photocatalytic Oxidation of Methane with Water
Article Publication Date
27-Jun-2023