Credit: POSTECH
Predicting the impact of DNA sequence variants is important for sorting disease-associated variants (DVs) from neutral variants. Korean researchers at Pohang University of science and technology (POSTECH) report the development of a method to predict the impact of DVs. The study appears in the journal Nucleic Acids Research in June.
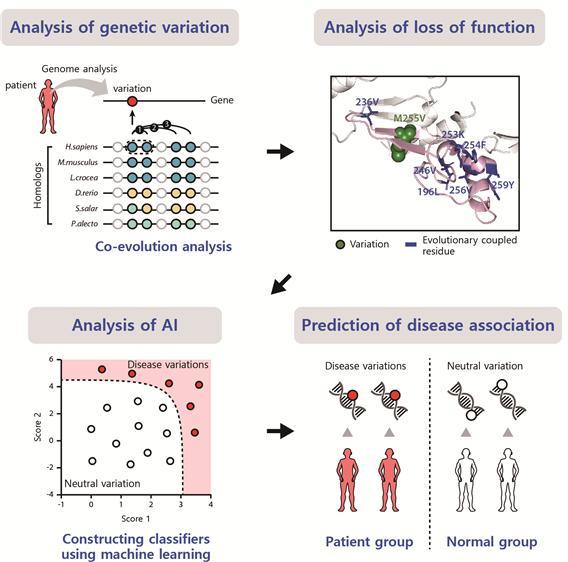
Current methods to predict the mutational impacts depend on evolutionary conservation at the mutation site, which is determined using homologous sequences and based on the assumption that variants at well-conserved sites have high impacts. However, many DVs at less-conserved but functionally important sites cannot be predicted by the current methods.
The researchers present a method to find DVs at less-conserved sites by predicting the mutational impacts using evolutionary coupling analysis. Functionally important and evolutionarily coupled sites often have compensatory variants on cooperative sites to avoid loss of function. They identified DVs at less-conserved sites that were not identified using current conservation-based methods.
Prof. Kim said that “This study can be applied to a variety of precision medicine approaches such as prognosis of patient’s diseases and finding personalized medicine.” “Based on a large scale sequence analysis, the developed method is useful to find more disease associated variants which help to find biomarkers and therapeutic targets of various human diseases.”
###
Media Contact
Hyeyong Choi
[email protected]
Original Source
http://www.
Related Journal Article
http://dx.



{kind=link}