A new tool created to simplify the comparison of RNA-Seq data
Credit: Courtesy of Nicolas Borisov
Scientists at Sechenov University, together with their Russian and American colleagues, developed the tool named Shambhala, which can process the “decoding” of RNA obtained on different equipment to make them suitable for comparison and further study. More information can be found in the BMC Bioinformatics journal.
To better understand how various diseases and drugs affect the cells, scientists study the structure of RNA, which is synthesized on the basis of DNA and reflects the activity (expression) of genes.
For more than 20 years of existence of such technologies, scientists have created several large databases of all known diseases. However, the obtained data can be compared only after their processing since different methods and equipment are used to analyze the composition of RNA, but the results may differ, even if they were obtained under the same conditions, but at different times.
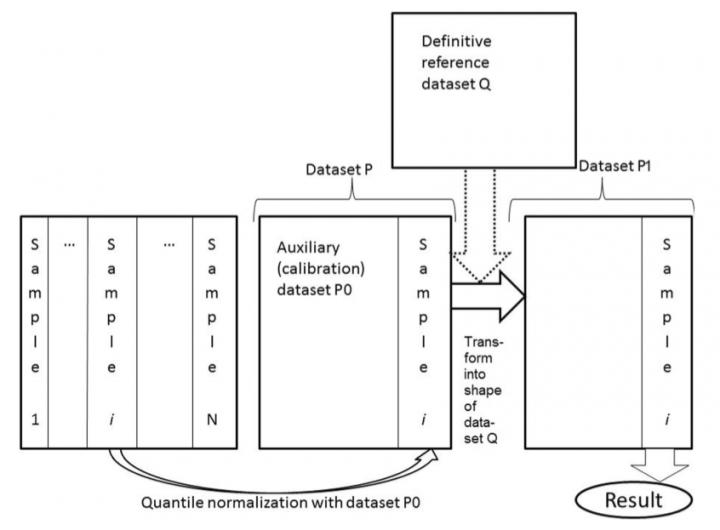
Unlike previously published methods enabling good quality data harmonization for only two datasets, Shambhala allows conversion of multiple datasets into the universal form suitable for further comparisons. The result of the work of the Affymetrix Human Gene microarray was chosen as such as it has the biggest number of human gene expression profiles deposited in public databases.
The Shambhala harmonization protocol includes several specific features such as the auxiliary calibration dataset that helps to initially transform the data, and the reference definitive dataset that defines the universal shape of the output harmonized gene expression profile. Each expression profile is calibrated independently, not as part of the initial dataset, which ensures the statistical stability of the harmonization result. The calibration dataset was selected to preserve important samples characteristics.
In order to test the code module, four biological samples (A, B, C, D) were analyzed using several experimental platforms, than Shambhala processed the results. The obtained data were clustered, i.e. divided into groups, to see how this division reflects the sample data.
The test confirmed that after Shambhala processing, clustering separates the data sets by biological attributes, rather than by platform used for sequencing. The clusters of biologically close samples (A and B, C and D) were located at a smaller distance from each other. However, the algorithm did not succeed in hierarchical clustering of such similar patterns. The efficiency varied depending on the selected final format and the reference definitive dataset.
Two other methods (Quantile Normalization and based on the binomial distribution DESeq / DESeq2, widely used for data harmonization) undergone the same test and showed worse results providing purely platform-specific outputs.
“The new method of harmonization of omics data made it possible to compare millions of expression profiles obtained from various samples of biological organs and tissues,” says the coauthor of the article Ni?olas Borisov, chief researcher, Institute of Personalized Medicine, Sechenov University.
###
The research conducted in collaboration with scientists from Petrozavodsk State University, Shemyakin-Ovchinnikov Institute of Bioorganic Chemistry, Institute of General Pathology and Pathophysiology, JSC Generium and OmicsWay Corp.
Media Contact
Nataliya Rusanova
[email protected]
Related Journal Article
http://dx.



{kind=link}