
The rapid advancement of single-cell RNA sequencing (scRNA-seq) technology has revolutionized biological research, offering unprecedented resolution to examine gene expression patterns at the level of individual cells. This extraordinary capability has facilitated extraordinary insights into cellular heterogeneity across diverse tissues and organisms, driving progress in fields such as immunology, oncology, and developmental biology. Despite over 40,000 studies utilizing scRNA-seq to map cellular diversity, researchers continue to grapple with a fundamental challenge: the instability and unreliability of clustering algorithms used to categorize cells based on their gene expression profiles.
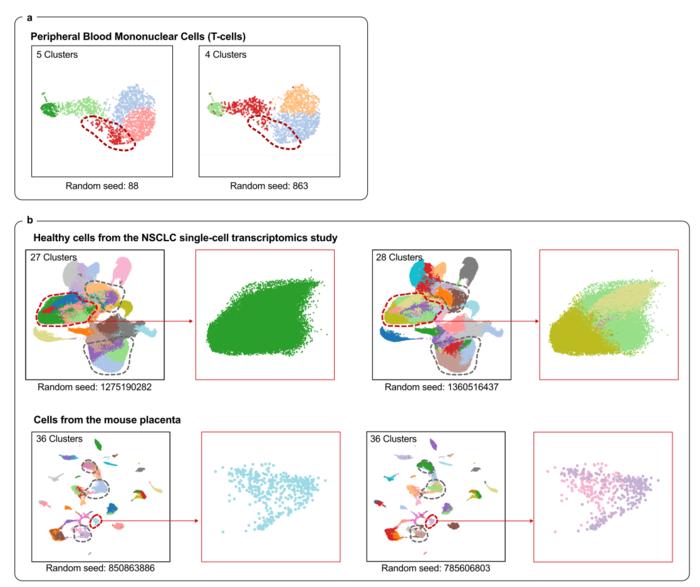
Clustering is a pivotal computational step in single-cell analysis, as it groups cells with similar gene expression patterns to identify cell types, states, and subpopulations. However, subtle variations in clustering parameters—like random seeds—can profoundly influence clustering outcomes, even when the same data is analyzed multiple times. This inconsistency creates a “reliability crisis” that undermines the biological interpretations drawn from scRNA-seq data and hinders clinical and therapeutic applications that depend on accurate cellular classification.
Misclassification can have serious consequences. For example, normal cells might be erroneously labeled as malignant, or rare but biologically crucial cell populations may be overlooked entirely. To address this, researchers have traditionally relied on consensus clustering methods that repeatedly assess whether pairs of cells are assigned to the same clusters across multiple runs. Although effective in principle, consensus clustering is computationally expensive and not scalable to the massive datasets produced by modern high-throughput scRNA-seq experiments, which often contain tens of thousands to hundreds of thousands of cells.
.adsslot_MQbunLKxWt{width:728px !important;height:90px !important;}
@media(max-width:1199px){ .adsslot_MQbunLKxWt{width:468px !important;height:60px !important;}
}
@media(max-width:767px){ .adsslot_MQbunLKxWt{width:320px !important;height:50px !important;}
}
ADVERTISEMENT
In response to these challenges, a team led by Professor Kim Jae Kyoung at the Korea Advanced Institute of Science and Technology (KAIST) and the Institute for Basic Science (IBS) has introduced scICE, a mathematically grounded framework designed to enhance the reliability and efficiency of clustering single-cell data. Published in Nature Communications, this study presents an innovative approach that sidesteps the computational bottlenecks associated with traditional consensus clustering and offers an automated way to evaluate clustering stability without exhaustive pairwise comparisons.
The cornerstone of scICE is its Inconsistency Coefficient (IC), a robust statistical metric that quantifies the stability of cell cluster assignments directly. By applying this measure, scICE identifies and filters out unstable cell groupings, preserving only those clusters that consistently represent true biological signals. This framework not only reduces computational complexity but also allows researchers to trust clustering results with greater confidence, facilitating downstream analyses and hypothesis testing.
Dr. Kim Hyun, the lead author from IBS, emphasizes the significance of this advance: “The reliability of single-cell clustering has been underappreciated, despite its critical importance for biological interpretation. scICE introduces a new paradigm for rapidly verifying clustering results, enabling researchers to proceed with greater certainty.” The approach fundamentally transforms how stability is assessed, improving both speed and accuracy.
To rigorously evaluate scICE’s performance, the team applied their framework to 48 diverse scRNA-seq datasets derived from both experimental and simulated sources, covering multiple tissues such as the brain, lungs, and blood. The findings were striking: approximately two-thirds of existing clustering results in these datasets were statistically unstable, revealing a pervasive issue of unreliability in commonly used approaches. In contrast, scICE effectively selected a smaller subset of highly reliable clusters, demonstrating exceptional precision while conserving computational resources.
The benefits of scICE extend beyond reliability alone. Notably, the framework exhibits a pronounced ability to detect rare cell populations—an area where conventional clustering methods frequently falter. Rare cell types often play essential roles in immune responses and disease processes, yet their identification is notoriously difficult due to their scarcity and the noise inherent in single-cell data. By facilitating subclustering informed by the Inconsistency Coefficient, scICE can illuminate these hidden populations, offering vital insights into cellular diversity.
Professor Kim Jae Kyoung highlights the practical impact of this innovation: “scICE empowers scientists to streamline their analytical pipelines by focusing on trustworthy clusters. We anticipate it will become an indispensable tool for the life sciences community, setting a new standard for the interpretation of single-cell RNA sequencing data.” The team’s commitment to open science is reflected in their decision to release scICE publicly on GitHub, fostering widespread adoption and enabling further improvement by the research community.
As single-cell technologies continue to generate ever more complex datasets, the need for reliable, scalable analytical tools becomes increasingly critical. scICE’s mathematical framework—rooted in the Inconsistency Coefficient—addresses this demand with elegance and efficiency. By ensuring reproducibility and accuracy of clustering results, scICE accelerates biological discovery and aids in translating single-cell insights into clinical advances.
This breakthrough underscores a broader imperative in computational biology: rigorous validation of analytical methods alongside data generation. As scRNA-seq and other omics techniques push the boundaries of resolution, sophisticated statistical tools like scICE will be essential for separating meaningful biological signals from technical noise and computational artifacts. Ultimately, these developments will deepen our understanding of cellular complexity, improve disease diagnosis, and inform the design of targeted therapies.
The research presented by Professor Kim and colleagues exemplifies the integration of mathematical innovation with biological inquiry, offering a powerful solution to a longstanding problem in single-cell science. By enhancing clustering reliability and computational efficiency, scICE promises to reshape the landscape of single-cell RNA sequencing analysis and inspire future methodological advancements in the field.
Subject of Research: Cells
Article Title: scICE: Enhancing Clustering Reliability and Efficiency of Single-cell RNA Sequencing Data with Multi-Cluster Label Consistency Evaluation
News Publication Date: 2-Jul-2025
Web References:
DOI link: 10.1038/s41467-025-60702-8
Image Credits: Institute for Basic Science
Keywords: Single cell sequencing, Genome sequencing strategies, Genomics, Genetics, Mathematical biology, Computational biology, Bioinformatics, Sequence analysis, Cluster analysis, Data analysis, Information processing, Immune cells, Cells, Cell biology, Developmental biology, Life sciences, Systems biology, Bioengineering, Engineering, Mathematical modeling, Applied mathematics
Tags: accuracy in cellular classificationaddressing instability in clustering outcomesadvancements in immunology researchcellular heterogeneity in biological researchclustering algorithms in single-cell analysisconsensus clustering methods for cell categorizationdevelopmental biology and single-cell studiesimplications of single-cell analysis in oncologyinnovative tools for single-cell datamisclassification in gene expression profilingscRNA-seq data reliability challengessingle-cell RNA sequencing technology



{kind=link}