LA JOLLA, CA—Genetic mutations kick start cancers. Some mutations shuffle the genetic code, others come from the deletion of key genes.
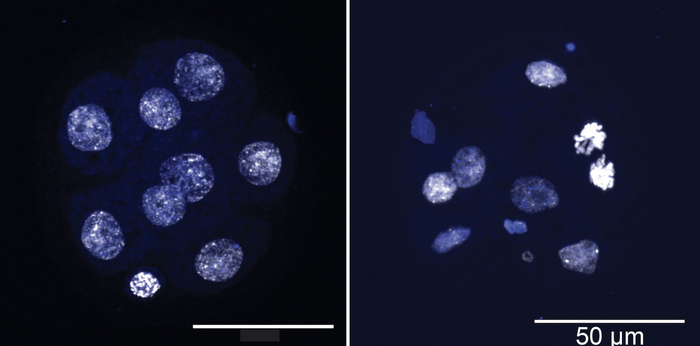
Credit: Image courtesy Rao Lab, La Jolla Institute for Immunology
LA JOLLA, CA—Genetic mutations kick start cancers. Some mutations shuffle the genetic code, others come from the deletion of key genes.
At La Jolla Institute for Immunology (LJI), researchers have made a major breakthrough in understanding how deletion of the genes that encode TET proteins can lead to cancer growth. Their new study, published in Nature Communications, is the first to show the immediate consequences of deleting all three genes from the TET family in mouse embryonic stem cells.
By using this mouse model, the researchers discovered that TET proteins are critical for keeping the process of cell and DNA replication running smoothly. Without TET proteins, important genes go missing, leading to the mutations, or aneuploidies (an-new-ploy-dees).
Aneuploidies are cases where genetic material is added or deleted on a massive scale. Cells with aneuploidies aren’t just missing a gene. Instead, genes go missing across an entire chromosome. “Aneuploidies are a common feature of cancer cells,” says LJI Postdoctoral Researcher Hugo Sepulveda, Ph.D.
Uncovering this direct connection between TET loss of function and aneuploidies is a major discovery in the field of cell biology, and it gives researchers a clue to how to find genes that underpin cancer development. “We can now understand the mechanisms behind aneuploidy development, although we can’t say these changes always happen through the same genes in other cell types,” says LJI Postdoctoral Researcher Hugo Sepulveda, Ph.D.
Sepulveda co-led the research with former LJI postdoctoral researcher Romain Georges, Ph.D., who generated the mouse model and derived the stem cells for the project. LJI Professor Anjana Rao, Ph.D., served as the study’s senior author.
What are TET proteins?
As a researcher at Harvard, Rao discovered the TET family of proteins with Mamta Tahiliani, Ph.D., and L. Aravind, Ph.D. Her work since then has shown that TET proteins are key players in cell growth and development. TET proteins can protect against cancer-causing mutations and even guard against inflammation and cardiovascular disease. TET proteins have such an important role in cells because they influence DNA methylation, a process that alters how DNA is read and genes are expressed.
Rao’s work was especially important for understanding TET function in immune cells, such as T cells, B cells, and myeloid cells. “Dr. Rao showed that every time you have a deletion of a TET gene in these cells, you see the development of a different aggressive type of cancer,” says Sepulveda.
As this research continued, the LJI team noticed something odd—cells with missing or impaired TET proteins also tend to have aneuploidies. Here was another link between TET proteins and cancers.
Cells with TET loss of function tended to have aneuploidies, and cancer cells tended to have aneuploidies. But what comes first? Does TET loss of function trigger aneuploidies and cancers, or is it the other way around?
An exciting discovery
To better understand cancer, Georges and Sepulveda turned to mouse embryonic stem cells as a model. These cells were naturally poised to divide rapidly but not prone to developing cancers. The researchers needed to see how deleting TET proteins could shake things up.
Again and again, Georges, Sepulveda, and their colleagues found that cells with TET deletion developed aneuploidies at three times the rate of normal cells. These altered cells lost genes very quickly and randomly. The scientists could see the effects in very early embryos, which consisted of just eight cells.
“That proved that TET deletion had a direct effect on aneuploidies,” Sepulveda says. “That was very exciting and had not been shown before.”
Next, the researchers turned to a sequencing technique called RNA-seq to see how TET deletion affects other genes. They saw a “downregulation,” or switching off, of certain genes associated with cell and DNA replication. This finding suggested that TET deletion was a major blow to a system that keeps cells dividing normally.
So which genes are to blame?
TET deletion in mouse embryonic stem cells appears to have the greatest effect on a gene called Khdc3, which belonged to a system, or complex, that was previously studied for its activity in helping oocytes divide. This complex is not well studied, but Khdc3 was known to be important for maintaining genome stability in oocytes before and after fertilization, and in the early stages of embryonic development.
When the researchers restored KHDC3 protein function in these cells, they were surprised to see that genome stability returned as well. Aneuploidy was reversed. The complex that Khdc3 is part of was back to doing its job.
The new study revealed two key facts regarding TET loss of function. First, that TET loss of function is a direct cause of the aneuploidies linked to cancers, since it resulted in decreased Khdc3 expression. Second, that TET loss of function in embryonic stem cells affects genome stability via some KHDC3-containing complex.
Sepulveda is careful to note that the Khdc3 complex is only known to be active in early embryonic development and in embryonic stem cells. This means that even if aneuploidies are seen in TET-deficient cancers, scientists till need to determine whether these cancers upregulate KHDC3 (most cancers tend to upregulate embryonic genes) and if so, whether the aneuploidies they develop are caused by aberrant KHDC3 function.
Notably, aneuploidies are seen in numerous cancers in which TETs are not mutated, but these cancers could have lost TET function because of metabolic disturbances.
“Genome instability in cancer cells could be happening through genes other than Khdc3, but through a similar regulatory mechanism that also involves changes on DNA methylation patterns,” says Sepulveda. “Whether TET-associated cancers develop aneuploidies by dysregulating different genes than Khdc3 is still an open question.”
Going forward, Sepulveda hopes to uncover exactly how the Khdc3 complex promotes genome stability downstream of TET proteins in embryonic stem cells.
Additional authors of the study, “Acute deletion of TET enzymes results in aneuploidy in mouse embryonic stem cells through decreased expression of Khdc3,” include Carlos Angel, Eric Johnson, Susan Palomino, Roberta Nowak, Arshad Desai, and Isaac F. López-Moyado.
This work was supported by the National Institutes of Health (grants R35 CA210043, R01 GM074215, NIH S10OD016262, NIH S10RR027366, AI125179, and F32 postdoctoral fellowships), the Pew Latin-American Fellows Program from The Pew Charitable Trusts, the University of California Institute for Mexico and the United States–Consejo Nacional de Ciencia y Tecnología (UC MEXUS-CONACYT) Fellowship.
This research used resources of the Advanced Light Source, which is a DOE Office of Science User Facility under contract no. DE-AC02-05CH11231. The NovaSeq 6000 and the HiSeq 2500 were acquired through the Shared Instrumentation Grant (SIG) Program (S10); NovaSeq 6000 S10OD025052 and HiSeq 2500 S10OD016262.
DOI: 10.1038/s41467-022-33742-7
Journal
Nature Communications
DOI
10.1038/s41467-022-33742-7
Method of Research
Experimental study
Subject of Research
Cells
Article Title
Acute deletion of TET enzymes results in aneuploidy in mouse embryonic stem cells through decreased expression of Khdc3
Article Publication Date
20-Oct-2022
COI Statement
No COI

{kind=link}