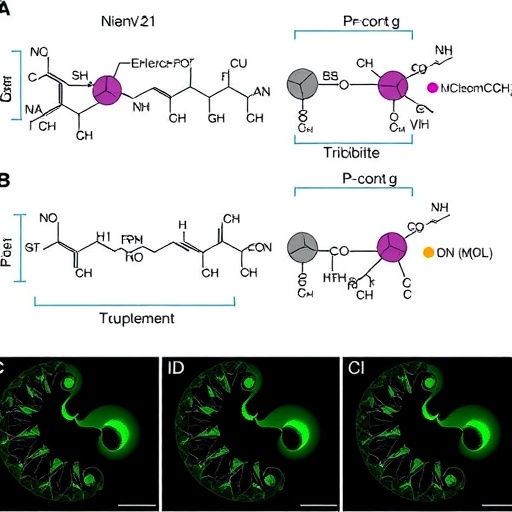
In a groundbreaking study published in Molecular Diversity, researchers Xu, S., Yang, Y., and Chen, C. have harnessed the power of Bayesian active learning to facilitate structure-based virtual screening, identifying novel inhibitors of mutant IDH1. This innovative approach combines advanced computational techniques with a deep understanding of molecular interactions, leading to significant strides in targeted cancer therapies.
Mutant IDH1 is known to play a pivotal role in various cancers, particularly gliomas and acute myeloid leukemia. It is an enzyme that is crucial for cellular metabolism, and its mutations alter metabolic pathways, promoting oncogenesis. The ability to inhibit mutant IDH1 effectively is, therefore, of great importance to clinicians and researchers aiming to develop more effective cancer treatments. By targeting this mutation, therapeutic strategies can be personalized and potentially more effective in eradicating cancer cells.
The research team employed a structure-based virtual screening method enhanced by Bayesian active learning, which optimizes the selection of compounds based on their predicted activity towards the target protein. This innovative methodology not only accelerates the discovery process but also ensures that the most promising candidates are prioritized for further investigation. Unlike traditional high-throughput screening methods, this approach allows for a more nuanced understanding of how different compounds interact with mutant IDH1.
To achieve their objectives, the researchers created a comprehensive model of the mutant IDH1 structure. This computational model serves as a framework for identifying potential inhibitor candidates through virtual docking simulations. By simulating how various compounds interact with the mutated enzyme, the team could predict which inhibitors would bind effectively, paving the way for the discovery of novel therapeutic agents.
One of the pivotal components of their research was the integration of Bayesian inference, which aids in refining predictions based on experimental outcomes. As the team conducted virtual screenings and synthesized experimental results, their predictive model continually evolved, improving its accuracy over time. This loop of prediction and validation exemplifies a significant advance in computational drug discovery, offering a more dynamic and responsive system for identifying potential therapeutic agents.
With initial candidates identified, the researchers proceeded to validate their virtual screening findings through laboratory experiments. These experiments included biochemical assays to measure the efficacy of the proposed inhibitors against mutant IDH1. Preliminary findings indicated promising results, with several compounds demonstrating potent inhibitory effects, thus validating the computational predictions made during the screening.
The implications of these findings extend beyond just the identification of new inhibitors. The study exemplifies a paradigm shift in drug discovery, where computational methods and machine learning techniques are increasingly integral to the drug development process. The integration of such technologies facilitates not just the identification of potential therapeutics but also allows researchers to better understand the mechanisms of action and potential side effects associated with these compounds.
As researchers continue to optimize and expand upon this methodology, the potential for rapid advancements in cancer therapy becomes more tangible. The ability to quickly identify and confirm the efficacy of drug candidates could dramatically shorten the timeline for bringing new cancer therapies to the clinic. This study provides a glimpse into the future of oncology, where personalized medicine becomes a reality through the application of cutting-edge technology.
Building upon this work, researchers are already discussing the potential for collaborations across various institutions and disciplines to further enhance their methodologies. By sharing datasets and pooling computational resources, the scientific community can accelerate the pace of discovery and maximize the impact of their findings on patient care. The collaborative nature of research in this field is essential to overcoming the significant challenges posed by cancer treatment.
Moreover, the findings could lead to a broader exploration of other mutations and cancer types, utilizing similar computational approaches to identify new therapeutic avenues. This flexibility underscores the versatility of the Bayesian active learning framework, which could be adapted for various applications within drug discovery and development.
The broader implications of this research extend to our understanding of cancer biology and the role of metabolic dysfunction in tumorigenesis. As researchers delve deeper into the metabolic pathways influenced by mutant IDH1, they uncover potential biomarkers for early detection and prognosis, enabling clinicians to tailor treatment strategies more effectively.
In conclusion, the research conducted by Xu, S., Yang, Y., and Chen, C. marks a significant advancement in the field of cancer therapeutics. Their innovative use of Bayesian active learning and structure-based virtual screening not only sheds light on the intricacies of mutant IDH1 but also paves the way for future discoveries that could revolutionize cancer treatment. As we stand on the brink of a new era in personalized medicine, these findings serve as a testament to the power of computational approaches in driving scientific progress.
As scientists continue to explore the vast potential of this approach, the hope of developing effective treatments for cancers associated with mutant IDH1 has never been more promising. By marrying computational prowess with experimental validation, the future of cancer therapies looks brighter, with the possibility of more targeted, effective, and personalized options for patients on the horizon.
Subject of Research: Novel inhibitors of mutant IDH1
Article Title: Bayesian active learning-aided structure-based virtual screening reveals novel inhibitors of mutant IDH1
Article References:
Xu, S., Yang, Y., Chen, C. et al. Bayesian active learning-aided structure-based virtual screening reveals novel inhibitors of mutant IDH1.
Mol Divers (2025). https://doi.org/10.1007/s11030-025-11381-6
Image Credits: AI Generated
DOI: 10.1007/s11030-025-11381-6
Keywords: Bayesian active learning, structure-based virtual screening, mutant IDH1, cancer therapy, drug discovery, metabolic pathways.
Tags: advanced screening methods for cancer therapeuticsBayesian active learning in drug discoverycomputational methods in cancer therapyenzyme inhibitors in oncologygliomas and acute myeloid leukemia researchinnovative approaches to targeted cancer therapiesmolecular interactions in drug designmutant IDH1 targeting in cancernovel IDH1 inhibitorsoptimizing compound selection for drug developmentpersonalized cancer treatment strategiesstructure-based virtual screening techniques

{kind=link}