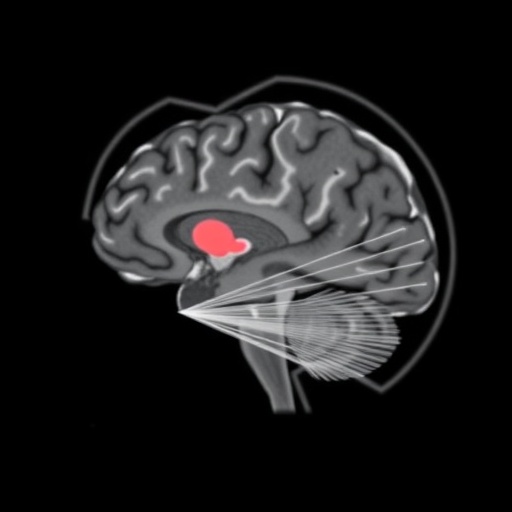
In a groundbreaking study conducted at the VIB-UAntwerp Center for Molecular Neurology, researchers have unveiled novel insights into the development of brain networks in a mouse model of KCNQ2-related developmental and epileptic encephalopathy (KCNQ2-DEE). This rare but devastating neurological disorder primarily afflicts neonates, manifesting initial seizures mere days after birth, followed by enduring challenges in cognition and motor functions. By leveraging advanced longitudinal imaging technologies, the team meticulously tracked changes in brain connectivity patterns over time, revealing disruptions in neural communication that precede overt behavioral symptoms by a significant margin.
KCNQ2-DEE stems from mutations in the KCNQ2 gene, which encodes a critical potassium ion channel pivotal for maintaining the brain’s electrical stability. These mutations undermine the normal gating of potassium ions across neuronal membranes, leading to pathological hyperexcitability and seizure activity. Despite the gene’s well-established role in modulating neuronal excitability, the comprehensive effects of such mutations on the intricacies of brain circuit maturation have remained elusive. To address this gap, Professor Sarah Weckhuysen and her team engineered a murine model harboring the human-equivalent KCNQ2 mutation, enabling detailed functional and structural brain investigations during critical early developmental windows.
Central to the study’s methodology was the application of non-invasive neuroimaging modalities, including magnetic resonance imaging (MRI) and positron emission tomography (PET). These techniques allowed for the in vivo appraisal of both anatomical structures and functional neural dynamics across multiple timepoints. Intriguingly, the researchers observed that despite the profound neurological consequences of the mutation, gross brain morphology remained largely preserved. Instead, it was the functional connectivity—the dynamic communication between distinct brain regions—that exhibited marked alterations. Such findings underscore a critical distinction: the disorder’s primary pathology resides within the operational dynamics of the brain’s circuitry rather than in its physical architecture.
This functional disconnect echoes patterns identified in a constellation of other neurodevelopmental disorders, notably autism spectrum disorder and schizophrenia, where disruption of ion channel function has been implicated in aberrant brain network maturation. The parallels suggest a convergent mechanism by which ion channelopathies derail the fine-tuning of synaptic connections critical for neurodevelopment. These early disruptions in connectivity may lay the groundwork for the varied neurological manifestations observed clinically, including seizures, cognitive deficits, and motor dysfunctions.
Remarkably, the team’s longitudinal approach revealed that these aberrant connectivity signatures emerge well before any discernible behavioral phenotypes, such as seizures or developmental delays, are evident. This temporal dissociation provides a valuable window for early diagnosis and potentially preemptive therapeutic intervention. The findings shift the paradigm from reactive symptomatic treatment to a proactive strategy focused on preserving and restoring network integrity during formative stages of brain development.
Professor Weckhuysen emphasized the clinical significance of these discoveries, noting that visualizing the trajectory of brain network development effectively illuminates the pathogenesis of KCNQ2-DEE. Such insights can pinpoint critical periods and neural substrates most vulnerable to therapeutic modulation, guiding the optimization of intervention timing and modalities. Early treatment, she suggests, could transcend seizure control alone, potentially correcting or mitigating broader deficits arising from disrupted circuit maturation.
The research team’s investigative trajectory has not been limited to descriptive studies. Prior explorations from the Weckhuysen lab identified an existing antipsychotic compound capable of modulating potassium channel activity. This pharmacological agent, initially characterized for other indications, may offer a promising candidate for repurposing as a targeted therapy for KCNQ2-DEE. The elucidation of when and how circuit disruptions manifest will be instrumental in assessing the efficacy and timing of such treatments.
Moreover, the study highlights a broader principle within neurobiology: that ion channel mutations’ impact extends beyond simple electrical disturbances to fundamentally alter developmental programming of brain networks. This recognition advocates for integrative approaches combining genetics, molecular neuroscience, and systems-level imaging to unravel the complex pathophysiology of rare neurological disorders.
Future research will likely build upon these findings to refine biomarkers capable of detecting subtle functional brain changes in human patients carrying KCNQ2 mutations. Early identification of at-risk infants could pave the way for bespoke therapeutic regimens tailored to individual developmental trajectories. Additionally, longitudinal neuroimaging could serve as an invaluable tool for monitoring treatment response and disease progression.
This study represents a significant stride toward understanding the temporal and mechanistic underpinnings of KCNQ2-DEE. By redefining the disorder as a condition of disrupted brain network maturation rather than purely structural anomalies, it opens novel avenues for research and clinical application. Harnessing these insights promises to transform patient outcomes, shifting from management of symptoms to modification of disease courses at their earliest stages.
The implications of this work also extend beyond KCNQ2-DEE, offering a model to interrogate other ion channel-related neurodevelopmental disorders. It underscores the therapeutic potential embedded in early detection and intervention strategies aimed at restoring normative connectivity patterns in the developing brain. As the field advances, such integrative approaches will be essential to unraveling the complexities of brain maturation and its perturbations in disease.
In closing, Professor Weckhuysen calls for a renewed focus on elucidating the developmental trajectories disrupted by ion channelopathies, reaffirming the critical importance of early-stage research in translating molecular insights into clinical breakthroughs. Their results not only enhance our understanding of KCNQ2-related encephalopathies but also catalyze the search for effective interventions tailored to the unique needs of affected neonates and children.
Subject of Research: Animals
Article Title: Imaging brain development in a KCNQ2-developmental and epileptic encephalopathy mouse model: identifying early biomarkers for functional and structural brain changes
News Publication Date: 24-Oct-2025
Web References: http://dx.doi.org/10.1016/j.ebiom.2025.105986
Keywords: Neuroscience, Organismal biology, Signal transduction, Developmental biology, Molecular biology
Tags: advanced neuroimaging techniquesbrain function disruptions in neonateschildhood neurological disorderscognitive challenges in KCNQ2-DEEepilepsy in infantsKCNQ2-related developmental encephalopathylongitudinal imaging technologies in neurosciencemurine models in neurological researchneural communication and brain connectivitypotassium ion channel mutationsseizures in early childhoodunderstanding brain circuit maturation

{kind=link}