Credit: Peter Zwart, Berkeley Lab
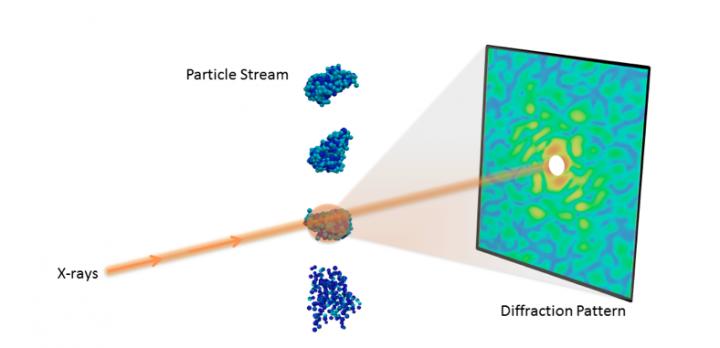
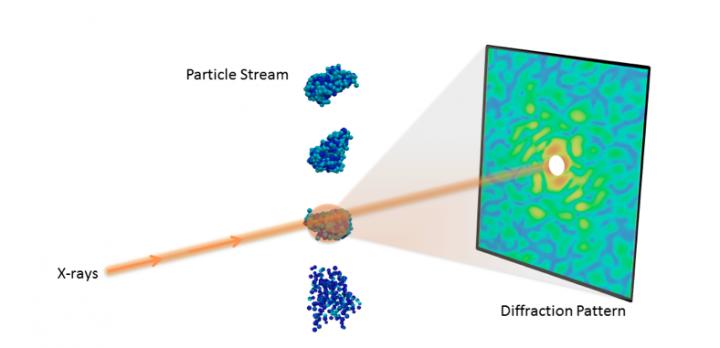
Understanding the 3D molecular structure of important nanoobjects like proteins and viruses is crucial in biology and medicine. With recent advances in X-ray technology, scientists can now collect diffraction images from individual particles, ultimately allowing researchers to visualize molecules at room temperature.
However, determining 3D structure from these single-particle diffraction experiments is a significant hurdle. For instance, current data acquisition rates are very limiting, typically resulting in fewer than 10 useful snapshots per minute, limiting the amount of features that can be resolved. Additionally, the images are often highly corrupted with noise and other experimental artefacts, making it difficult to properly interpret the data.
To meet these challenges, a team of researchers from the Lawrence Berkeley National Laboratory (Berkeley Lab) has developed a new algorithmic framework called multi-tiered iterative phasing (M-TIP) that utilizes advanced mathematical techniques to determine 3D molecular structure from very sparse sets of noisy, single-particle data. This approach essentially allows researchers to extract more information from experiments with limited data. Applied mathematicians Jeffrey Donatelli and James Sethian, and physical bioscientist Peter Zwart introduced this framework by expanding on an algorithm that they originally developed to solve the reconstruction from a related X-ray scattering experiment, called fluctuation X-ray scattering. A paper describing the M-TIP framework was published June 26 in the Proceedings of the National Academy of Sciences.
"This approach has the potential to revolutionize the field," says Zwart. "Given that it is hard to get a lot of good data, approaches that reduce the amount of data needed to successfully image 3D nanoobjects are likely to receive a warm welcome."
Donatelli, Sethian and Zwart are all part of CAMERA (The Center for Advanced Mathematics for Energy Research Applications), whose mission is to create the state-of-the-art mathematics required to handle data from many of DOE's most advanced scientific facilities. CAMERA is jointly funded by the Advanced Scientific Computing Research and Basic Energy Sciences programs in DOE's Office of Science.
Single Particle Diffraction
The recent advent of X-ray free-electron lasers (XFELs) has enabled several new experimental techniques for studying biomolecules that were infeasible with traditional light sources. One such technique is single-particle diffraction, which collects a large number of X-ray diffraction snapshots with only a single particle in the beam. By leveraging the extreme power of XFELs, researchers can collect measurable signals even from the tiniest particles.
One big advantage offered by this single-particle diffraction technique is the ability to study how different copies of a molecule vary or change in shape. Since each image comes from a single particle, these variations can be captured in the experiment, in contrast to traditional imaging methods like crystallography or small-angle X-ray scattering, where researchers can only measure an average over all different states of the molecular sample.
However, determining the 3D structure from single-particle diffraction data is challenging. To begin, when each particle is imaged, its orientation is unknown and needs to be recovered in order to properly combine the data into a 3D diffraction volume. This problem is compounded if the molecule can take on different shapes, which requires additional classification of the images. Furthermore, phase information is not recorded in diffraction images and must be recovered in order to complete the reconstruction. Finally, even with powerful XFELs, the number of scattered photons is very small, resulting in extremely noisy images, which can be further contaminated by systematic background and detector readout issues.
Previous approaches are based on solving the reconstruction problem in separate steps, where each individual problem is addressed separately. Unfortunately, a drawback to these serial approaches is that they do not easily leverage prior known features about what the molecule looks like. In addition, any error committed in one step is propagated to the next, resulting in a further increase in error. This "error snowball" ultimately degrades the quality of the reconstruction obtained in the final step.
Best of Both Worlds
Instead of solving the computational problems in separate steps, the team's M-TIP algorithm solves all parts of the problem concurrently. This approach leverages prior information about the structure to greatly reduce the degrees of freedom of the problem in all steps, and consequently reduce the required information needed to achieve a 3D reconstruction.
"Standard black-box optimization techniques can incorporate prior knowledge into the reconstruction but throw away all of the structure of the problem, whereas solving it in completely separate serial substeps exploits the structure of the problem but throws away almost all prior information about what the solution might look like," Donatelli said. "M-TIP leverages the best of both worlds by exploiting the structure of the problem to break up the computation into several manageable chunks and then iteratively refining over all of these chunks to arrive at a solution which is consistent with both the data and any structural constraints."
Using this technique, the team was able to determine 3D structure from extremely low image counts from simulated data, as low as 6 to 24 images for noise-free data and 192 images from highly contaminated data.
Breaking New Ground
This work is part of a new collaboration initiative between SLAC National Accelerator Laboratory, CAMERA, the National Energy Research Scientific Computing Center (NERSC) and Los Alamos National Laboratory as part of DOE's Exascale Computing Project (ECP). The goal of the project is to develop the computational tools necessary to perform real-time data analysis from experiments being conducted at SLAC's Linac Coherent Light Source (LCLS). With upgrades to the beamline, LCLS-II plans to generate several terabytes of data per second, which, for example, will allow scientists to greatly expand upon current single-particle experiments. Analyzing all of this data in real-time will require new algorithms and large computing machines. The M-TIP algorithm will serve as part of this process.
"These are some of the most challenging problems in computational data science," says Sethian. "To tackle them, we need to exploit a range of technologies, including emerging exascale computing architectures, sophisticated high speed networks, and the most advanced mathematical algorithms available. Bringing CAMERA scientists together with exascale application projects has opened the door to building tools to approach some pressing problems in biology and materials sciences."
The researchers note that these are just the first steps. In order for the method to be ready to be deployed, other hurdles have to be overcome.
"Experimental science is messy," says Zwart. "There are additional experimental effects that have to be taken into consideration in order for us to get the best possible results."
"Fortunately, M-TIP is a very modular technique," adds Donatelli, "so, it is well suited to modeling many of these additional effects without needing to change the core algorithmic framework."
The team is currently working on studying these effects as part of the Single Particle Initiative, a large, multi-institutional collaboration dedicated to addressing theoretical and practical issues in X-FEL-based single molecule imaging, ultimately leading to providing the scientific community with the tools needed to break new ground in biology, medicine and energy sciences.
###
NERSC is a DOE Office of Science User Facility at Berkeley Lab and LCLS is a DOE Office of Science User Facility at SLAC. The work was supported by the National Institute Of General Medical Sciences of the National Institutes of Health (NIH) and the DOE Office of Science, the single largest supporter of basic research in the physical sciences in the United States.
Media Contact
Linda Vu
[email protected]
510-495-2402
@BerkeleyLab
Original Source
http://cs.lbl.gov/news-media/news/2017/new-berkeley-lab-algorithms-extract-biological-structure-from-limited-data/


{kind=link}