Credit: Jeremy Goecks, Bassel F. El-Rayes, Shishir K. Maithel, H. Jean Khoury, James Taylor, Michael R. Rossi
There is an enormous amount that we do not understand about the fundamental causes and behavior of cancer cells, but at some level, experts believe that cancer must relate to DNA and the genome.
In their seminal 2011 paper, "The Hallmarks of Cancer: The Next Generation," biologists Douglas Hanahan and Robert Weinberg identified six hallmarks, or commonalities, shared by all cancer cells.
"Underlying these hallmarks are genome instability, which generates the genetic diversity that expedites their acquisition, and inflammation, which fosters multiple hallmark functions," they wrote.
An approach that has proved very successful in uncovering the complex nature of cancer is genomics — the branch of molecular biology concerned with the structure, function, evolution, and mapping of genomes.
Since the human genome consists of three billion base pairs, it is impossible for an individual to identify single mutations by sight. Hence, scientists use computing and scientific software to find connections in biological data. But genomics is more than simple pattern matching.
"When you move into multi-dimensional, structural, time-series, and population-level studies, the algorithms get a lot harder and they also tend to be more computationally intensive," said Matt Vaughn, Director of Life Sciences Computing at the Texas Advanced Computing Center (TACC). "This requires resources like those at TACC, which help large numbers of researchers explore the complexity of cancer genomes."
FISHING IN BIG DATA PONDS
A group led by Karen Vazquez, professor of pharmacology and toxicology at The University of Texas at Austin, has been working to find correlations between chromosomal rearrangements — one of the hallmarks of cancer genomes — and certain DNA sequences with the potential to fold into secondary structures.
These structures, including hairpin or cruciform shapes, triple or quadruple-stranded DNA, and other naturally-occurring, but alternative, forms, are collectively known as "potential non-B DNA structures" or PONDS.
PONDS enable genes to replicate and generate proteins and are therefore essential for human life. But scientists also suspect they may be linked to mutations that can elevate cancer risk.
Using the Stampede and Lonestar supercomputers at TACC, Vasquez worked with researchers from the University of Texas MD Anderson Cancer Center and Cardiff University to test the hypothesis that PONDS might be found at, or near, rearrangement breakpoints — locations on a chromosome where DNA might get deleted, inverted, or swapped around.
By analyzing the distribution of PONDS-forming sequences within about 1,000 bases of approximately 20,000 translocations and more than 40,000 deletion breakpoints in cancer genomes, they found a significant association between PONDS-forming sequences and cancer. They published their results in the July 2016 issue of Nucleic Acids Research.
"We found that short inverted repeats are indeed enriched at translocation breakpoints in human cancer genomes," said Vazquez.
The correlation recurred in different individuals and patient tumor samples. They concluded that PONDS-forming sequences represent an intrinsic risk factor for genomic rearrangements in cancer genomes.
"In many cases, translocations are what turn a normal cell into a cancer cell," said co-author Albino Bacolla, a research investigator in molecular and cellular oncology at MD Anderson. "What we found in our study was that the sites of chromosome breaks are not random along the DNA double helix; instead, they occur preferentially at specific locations. Cruciform structures in the DNA, built by the short, inverted repeats, mark the spots for chromosome breaks, mutations, and potentially initiate cancer development."
While the study provides evidence that PONDS-forming repeats promote genomic rearrangements in cancer genomes, it also raises new questions, such as why PONDS are more strongly associated with translocation than with deletions?
Vasquez and her collaborators have followed up their computational research with laboratory experiments that explore the specific conditions under which translocations form cancer-inducing defects. Writing in Nucleic Acids Research in May 2017, she described how a specific 23-base pair-long translocation breakpoint can form a potential non-B DNA structure known as H-DNA, in the presence of sodium and magnesium ions.
"The predominance of H-DNA implicates this structure in the instability associated with the human c-MYC oncogene," Vasquez and her collaborators wrote.
Understanding the processes by which PONDS lead to chromosomal rearrangements, and these rearrangements impact cancer, will be important for future diagnostic and treatment purposes.
[The National Cancer Institute, part of the National Institutes of Health, funded these studies.]
ANALYZING THE GENOME IN ACTION
With the exception of mutations, the genome remains roughly fixed for a given cell line. On the other hand, the transcriptome — the set of all messenger RNA molecules in one cell or a population of cells — can vary with external conditions.
Messenger RNA (mRNA) convey genetic information from DNA to the ribosome, where they specify what proteins the cell should make — a process known as gene expression. Understanding what genes are being expressed in a tumor helps to more precisely classify tumors into subgroups so they can be properly treated.
Vishy Iyer, a professor of molecular biosciences at The University of Texas at Austin, has developed a way to identify sections of DNA that correlate with variations in specific traits, as well as epigenetic, or non-DNA related, factors that impact gene expression levels.
He and his group use this approach on data from The Cancer Genome Atlas (TCGA) to study the effects of genetic variation and mutations on gene expression in tumors. TACC's Stampede supercomputer helps them mine petabytes of data from TCGA to identify genetic variants and subtle correlations that relate to various forms of cancer.
"TACC has been vital to our analysis of cancer genomics data, both for providing the necessary computational power and the security needed for handling sensitive patient genomic datasets," Iyer said.
In February 2016, Iyer and a team of researchers from UT Austin and MD Anderson Cancer Center, reported in Nature Communications on a genome-wide transcriptome analysis of the two types of cells that make up the prostate gland — prostatic basal and luminal epithelial populations. They studied the cells' gene expression in healthy individuals as well as individuals with cancer, and identified cell-type-specific gene signatures that were associated with aggressive subtypes of prostate cancer that showed adverse clinical responses.
"By analyzing gene expression programs, we found that the basal cells in the human prostate showed a strong signature associated with cancer stem cells, which are the tumor originating cells," Iyer said. "This knowledge can be helpful in the development of more targeted therapies that seek to eliminate cancer at its origin."
Using a similar methodology, Iyer and a separate team of researchers from UT Austin and the National Cancer Institute identified a specific transcription factor associated with an aggressive type of lymphoma that is highly correlated with poor therapeutic outcomes. They published their results in the Proceedings of the National Academy of Sciences in January 2016.
By identifying these subtle indicators, not just in DNA but in mRNA expression, the work will help improve patient diagnoses and provide the proper treatment based on the specific cancers involved.
"Next-generation sequencing technology allows us to observe genomes and their activity in unprecedented detail," he said. "It's also making a lot of biomedical research increasingly computational, so it's great to have a resource like TACC available to us."
[These projects were supported, in part, by grants from NIH, DOD, Cancer Prevention Research Institute of Texas, MD Anderson Cancer Center Center for Cancer Epigenetics, Center for Cancer Research, Lymphoma Research Foundation and the Marie Betzner Morrow Centennial Endowment.]
POWERING CANCER RESEARCH THROUGH WEB PORTALS
With more than 30,000 biomedical researchers running more than 3,000 computing jobs a day, Galaxy represents one of the world's largest, most successful, web-based bioinformatics platforms.
Since 2014, TACC has powered the data analyses for a large percentage of Galaxy users, allowing researchers to quickly and seamlessly solve tough problems in cases where their personal computer or campus cluster is not sufficient.
Though Galaxy supports scientists studying a range of biomedical problems, a significant number use the platform to study cancer.
"Galaxy is like a Swiss army knife. You can run many different kinds of analyses, from text processing to identifying genomic mutations to quantifying gene expression and more," said Jeremy Goecks, Assistant Professor of Biomedical Engineering and Computational Biology at Oregon Health and Science University and one of the principal investigators for the project. "For cancer, Galaxy can be used to identify tumor mutations that drive cancer growth, find proteins that are overexpressed in a tumor, as well as for chemo-informatics and drug discovery."
He estimates that hundreds of researchers each year use the platform for cancer research, himself included. Because cancer patient data is closely protected, the bulk of this usage involves either publically available cancer data, or data on cancer cell lines – immortalized cells that reproduce in the lab and are used to study how cancer reacts to different drugs or conditions.
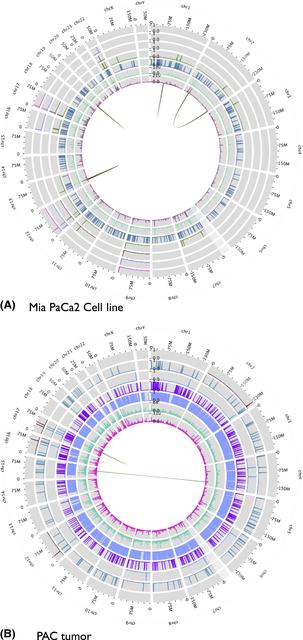
In Goecks's personal research, he develops data analysis pipelines to perform genomic profiles of pancreatic cancer and to use those profiles to find mutations associated with the disease and potentially useful drugs.
His work on exome and transcriptome tumor sequencing pipelines published in Cancer Research in January 2015, analyzed sequence data from six tumors and three common cell lines. He showed that they shared common mutations related to the KRAS gene, but that they also exhibited mutations not found in the cell lines, indicating the need to re-evaluate preclinical models of therapeutic response in the context of genomic medicine.
Broadly speaking, Galaxy helps researchers identify biomarkers that give an indication of a patient's prognosis and drug responses by placing individuals' genomic data in the context of larger cohorts of cancer patients, often from the International Cancer Genome Consortium or the Genomic Data Commons, both of which encompass more than 10,000 tumor genomes.
"Whenever you get a person's genomic data and a list of mutations which have arisen in the tumor but not in the rest of the body, the question is: 'Have we seen these mutations before?'" he explained. "That requires us to connect our individual patient data with these large cohorts, which tells us if we've seen it before and know how to treat it. This helps us determine if the cancer is aggressive or benign, or if we know particular drugs that will work given this particular mutation profile that the patient has."
The fact that it's now fast and inexpensive to generate DNA sequence data means lots of data is being produced, which in turn requires massive supercomputers like TACC's Stampede, Jetstream and Corral systems for analysis, storage and distribution.
"This is an ideal marriage of TACC having tremendous computing power with scalable architecture and Galaxy coming along and saying, 'we're going to go the last mile and make sure that people who can't normally use this hardware are able to.'"
As biology becomes an increasingly data-driven discipline, high-performance computing grows in importance as a critical component for the science.
"It's so easy to collect data from sequencing, proteomics, imaging. But when you have all of these datasets, you have to be able to process them automatically," he says. "The value of Galaxy is hiding some of the complexity that comes with that computing so that the scientist can focus on what matters to them: how to analyze a dataset to extract meaningful information, whether an analysis was successful, and how to produce knowledge by connecting analysis results with those in the broader biomedical community."
[The Galaxy Project is supported in part by NSF, NHGRI, The Huck Institutes of the Life Sciences, The Institute for CyberScience at Penn State, and Johns Hopkins University.]
###
Media Contact
Aaron Dubrow
[email protected]
512-471-8217
@TACC
http://www.tacc.utexas.edu/
Original Source
https://www.tacc.utexas.edu/-/mining-cancer-data-for-treatment-clues http://dx.doi.org/10.1093/nar/gkx100
############
Story Source: Materials provided by Scienmag


{kind=link}