A groundbreaking study has unveiled a pivotal role for macrophage-derived semaphorin 7A (SEMA7A) in the advancement of atherosclerosis, offering novel insights into the molecular mechanisms driving this chronic inflammatory disease. Atherosclerosis is characterized by the accumulation of lipid-rich foam cells within arterial walls, a pathological hallmark underlying major cardiovascular events such as myocardial infarction and stroke. Despite extensive research, treatments capable of halting or reversing plaque formation remain insufficient, prompting the search for innovative therapeutic targets. The recent investigation conducted by Fengchan Li and colleagues sheds light on the previously uncharted contribution of macrophage-specific SEMA7A in accelerating atherogenesis through an integrin β1/JNK/MSR1 signaling axis.
SEMA7A, traditionally recognized as an immunoregulatory molecule implicated in neuronal guidance and immune cell activation, has now been implicated in cardiovascular pathology with a macrophage-centered focus. Prior studies predominantly associated SEMA7A’s pro-atherogenic effects with vascular endothelium, but this novel research distinctly highlights macrophage-derived SEMA7A as a critical driver. Exploring macrophage-specific molecular pathways expands the horizon for understanding foam cell biology beyond conventional lipid accumulation theories, introducing immune regulation as a central element in plaque maturation and instability.
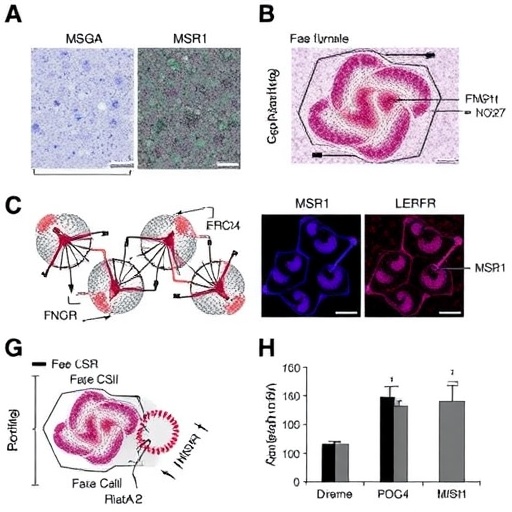
To elucidate the role of macrophage SEMA7A, the study began with comprehensive gene expression analyses using publicly available datasets. Human peripheral blood mononuclear cells demonstrated significantly elevated SEMA7A and integrin β1 expression in differentiated macrophages compared to monocytes, with protein levels increasing progressively during monocyte-to-macrophage maturation. This pattern underscores a differentiation-dependent regulatory mechanism wherein macrophages become primed to enhance SEMA7A-mediated signaling. Furthermore, exposure to oxidized low-density lipoprotein (ox-LDL), the quintessential atherogenic stimulus, synergistically augmented SEMA7A and integrin β1 expression, implicating a feedback loop that intertwines lipid accumulation with immunomodulatory pathways.
In vivo validation employed macrophage-restricted Sema7a knockout mice subjected to a well-established atherosclerosis model. Remarkably, deletion of SEMA7A in macrophages resulted in a dramatic 57.2% reduction in plaque burden compared to controls, accompanied by plaques indicative of greater stability, including diminished necrotic core size and thinner fibrous caps. These histological findings suggest a dual benefit: constraining lesion growth and mitigating the propensity for plaque rupture—a major precipitant of thrombotic events and clinical complications. Thus, macrophage SEMA7A emerges as a linchpin orchestrating both quantitative and qualitative aspects of atherosclerotic pathology.
Delving deeper into cellular mechanisms, the team uncovered that SEMA7A exerts its effects through direct interaction with integrin β1 receptors on macrophage surfaces, triggering activation of the c-Jun N-terminal kinase (JNK) pathway. The consequent downstream signaling cascade upregulates macrophage scavenger receptor 1 (MSR1), a major receptor responsible for internalizing modified lipoproteins. Heightened MSR1 expression facilitates excessive lipid uptake, leading to foam cell formation and accumulation within the arterial intima. This molecular axis—SEMA7A binding to integrin β1, JNK phosphorylation, and MSR1 induction—constitutes a robust feed-forward mechanism perpetuating atherosclerosis progression.
The therapeutic implications of targeting this newly identified molecular circuit were rigorously tested using GLPG0187, a potent antagonist of integrin β1 signaling. Administration of GLPG0187 to atherosclerotic mice significantly inhibited disease progression, substantiating the prospect of pharmacological intervention at the receptor level. This proof-of-concept paves the way for the development of targeted inhibitors that disrupt SEMA7A-integrin interactions or downstream pathways, potentially complementing existing lipid-lowering and anti-inflammatory therapies.
Beyond therapeutic avenues, the study enriches our fundamental understanding of atherosclerosis by situating macrophage SEMA7A at a critical nexus between immune regulation and lipid metabolism. It challenges researchers to reconsider the complex interplay between cellular adhesion molecules, kinase signaling, and scavenger receptors in the inflammatory milieu of vascular walls. Furthermore, the identification of SEMA7A as a biomarker correlated with ox-LDL exposure hints at its utility in cardiovascular risk stratification and monitoring treatment efficacy.
Future research is poised to explore the translational relevance of these findings within human clinical settings, including validation of SEMA7A and integrin β1 expression patterns in patient-derived atherosclerotic plaques. Investigations into combinatorial effects with statins or PCSK9 inhibitors may reveal synergistic benefits, optimizing therapeutic regimens. The pursuit of selective SEMA7A antagonists or integrin β1 modulators tailored for cardiovascular application represents a promising frontier, necessitating biochemical optimization and safety profiling.
Intriguingly, the cell-specific functions of SEMA7A across different vascular and immune compartments warrant detailed exploration to ascertain whether distinct mechanisms operate in smooth muscle cells, endothelial subsets, or other macrophage populations. Unraveling such spatial and functional heterogeneity could refine therapeutic targeting, minimizing off-target effects while maximizing disease attenuation. The complex signaling crosstalk within the arterial microenvironment underscores the necessity for systems-level analyses integrating transcriptomic, proteomic, and metabolomic data.
In conclusion, this seminal research delineates macrophage-derived SEMA7A as a master regulator of atherosclerosis via the integrin β1/JNK/MSR1 pathway. By unveiling a novel molecular axis driving foam cell formation and plaque progression, it provides transformative mechanistic insights with profound therapeutic potential. Targeting the SEMA7A signaling network holds promise to revolutionize cardiovascular disease management, offering hope for reducing the global burden of atherosclerosis-driven morbidity and mortality.
Subject of Research: Not applicable
Article Title: Macrophage-derived semaphorin 7A drives atherosclerosis via the integrin β1/JNK/MSR1 axis
News Publication Date: 15-Dec-2025
Web References: http://dx.doi.org/10.1007/s11684-025-1181-z
Image Credits: HIGHER EDUCATION PRESS
Keywords: Cell biology, Atherosclerosis, Macrophage, Semaphorin 7A, Integrin β1, JNK signaling, MSR1, Foam cells, Cardiovascular disease, Lipid uptake, Oxidized LDL, Therapeutic target
Tags: atherosclerosis molecular mechanismschronic inflammation in atherosclerosisfoam cell biology in plaque developmentimmune regulation in cardiovascular pathologyintegrin β1 signaling in atherogenesisJNK pathway in cardiovascular diseasemacrophage role in plaque instabilitymacrophage-derived semaphorin 7Amacrophage-specific therapeutic targetsMSR1 receptor in foam cell formationnovel targets for atherosclerosis treatmentsemaphorin 7A and immune cell activation


