Credit: @Vladimir Rybkin
The behavior of the solvated electron e-aq has fundamental implications for electrochemistry, photochemistry, high-energy chemistry, as well as for biology–its nonequilibrium precursor is responsible for radiation damage to DNA–and it has understandably been the topic of experimental and theoretical investigation for more than 50 years.
Though the hydrated electron appears to be simple–it is the smallest possible anion as well as the simplest reducing agent in chemistry–capturing its physics is…hard. They are short lived and generated in small quantities and so impossible to concentrate and isolate. Their structure is therefore impossible to capture with direct experimental observation such as diffraction methods or NMR. Theoretical modelling has turned out to be as challenging.
Density functional theory (DFT) is the electronic structure method most often used to study the solvated electron and water. Standard density functionals suffer from delocalization error though, making it impossible to model radicals accurately. Pure water complicates DFT approximations considerably, though choosing the right functionals can lead to acceptable results compared to high-level electronic structure benchmarks and values that can be observed through experiment. An accurate description of liquid water can be also achieved with many-body quantum chemistry methods, but they are extremely expensive.
Though a recent picosecond-scale molecular dynamics-based breakthrough unprecedented in complexity and requiring computational resources at the limits of what’s possible provided a crucial argument in favor of a cavity structure for e-aq, it did not result in other new insights or in a complete statistical description. Comprehensive characterization of the system’s properties requires far longer timescales, but simulating quantum nuclei at this level of electronic structure theory is currently beyond computational reach.
The modern way of working around this problem involves the use of machine learning. Training an ML force field or potential energy surface (PES) based on ab initio data allows for much longer MD simulations because the cost of evaluating such energies and forces is almost negligible compared to that associated with electronic structure calculations. The problem is that the solvated electron is a non-typical species. It doesn’t have an atomistic formula, which poses a problem because machine learning PES work with atomistic representations.
In the paper Simulating the Ghost: Quantum Dynamics of the Solvated Electron, University of Zurich researcher Vladimir Rybkin, doctoral student Jinggang Lan and lecturer Marcella Iannuzzi combined their expertise in electronic structure and solvated electrons with the knowledge of EPFL professor Michele Ceriotti and his former PhD students Venkat Kapil, now a researcher at Cambridge University, and Piero Gasparotto, now a researcher at Empa, in machine learning and quantum dynamics. That, with the contributions of other colleagues, resulted in the application of the ML approach to data acquired from a many-body quantum chemistry method known as second-order Møller-Plesset perturbation theory (MP2), a method that gives an accurate description of water, anyway, without any special treatment of the excess electron.
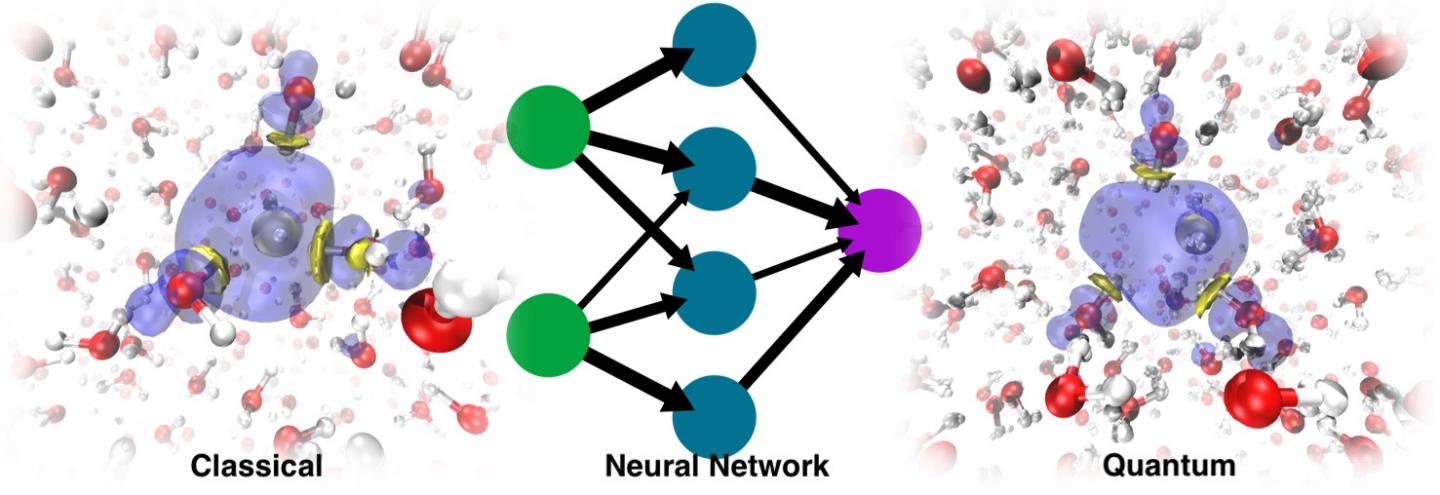
They were surprised to discover that the model was able to learn the presence of the solvated electron as a factor that distorted the structure of the pure liquid water. The dynamics run with the resulting ML PES was not only able to recover the stable cavity, but could also trace the correct localization dynamics, starting from the delocalized excess electron added to the water. In the end, ML simulated the electron as a sort of “ghost particle” that was not explicitly present in the model.
This allowed the researchers to achieve a time scale of several hundred picoseconds and collect reliable statistics by running a lot of computationally cheap classical trajectories and compute vibrational spectra, structures and diffusion. The ML approach also allowed them to simulate the quantum rather than classical nuclei with path-integral molecular dynamics (PIMD). This technique is at least one order of magnitude computationally more expensive than classical MD and cannot be carried out without ML PES at a high level of electronic structure theory.
Taking the nuclear quantum effects into account delivered accurate vibrational spectra, allowing the researchers to quantify the impact of these effects–already shown to be very important in the relaxation dynamics of the excess electron–on the hydrated electron. It also revealed transient diffusion, an unusual, rare event that is not present in the classical regime. While non-transient diffusion of the solvated electron is achieved by solvent exchange followed by gradual displacement of the “electron cloud” or spin density distribution, transient diffusion is rather a jump of the spin density from the stable cavity to the adjacent one.
While the ghost particle approach was applied here to the solvated electron, it could also be applied to excited states and quasiparticles such as polarons, opening up new opportunities for uniting high-level electronic structure theory with machine learning to achieve highly accurate dynamics simulations at a moderate price.
###
Media Contact
Carey Sargent
[email protected]
Original Source
https:/
Related Journal Article
http://dx.



{kind=link}