In the rapidly evolving world of genomics, single-cell technologies are at the forefront, unveiling intricate details about cellular behavior and the regulatory mechanisms that govern gene expression. Among these groundbreaking methodologies, single-cell assay for transposase-accessible chromatin using sequencing, or scATAC-seq, has dramatically transformed our understanding of chromatin accessibility. This technique allows for the exploration of the regulatory landscape of genomes at an unprecedented resolution, offering profound insights into gene regulation dynamics. However, as the application of scATAC-seq scales with mounting data from varied biological conditions, the pressing issue of batch effects comes into play, posing significant challenges for researchers aiming for accurate data interpretation.
Batch effects are systematic errors that arise when differences in sample processing, such as variations in laboratory conditions or sequencing runs, mask the inherent biological variations among the samples. This necessitates the need for robust data integration tools that can harmonize disparate datasets. While there has been considerable advancement in the field of single-cell RNA sequencing (scRNA-seq) integration, the existing tools fall short when applied to scATAC-seq data. The fundamental differences in data characteristics — such as sparsity in the measurement of accessible chromatin regions — restrict the effectiveness of traditional methods that have been developed primarily for transcriptomic data.
Existing integration approaches for scATAC-seq often compromise biological heterogeneity for the sake of adjusting for batch effects. Many of these techniques focus on low-dimensional corrections which, while addressing some aspects of batch variation, fail to preserve the invaluable biological information embedded within the data. This inadequacy can lead to distorted results that hinder downstream analyses, such as cell type identification or functional genomics exploration. Consequently, there has been an urgent need for new frameworks that can seamlessly integrate scATAC-seq datasets while maintaining the biological integrity of the underlying cellular compositions.
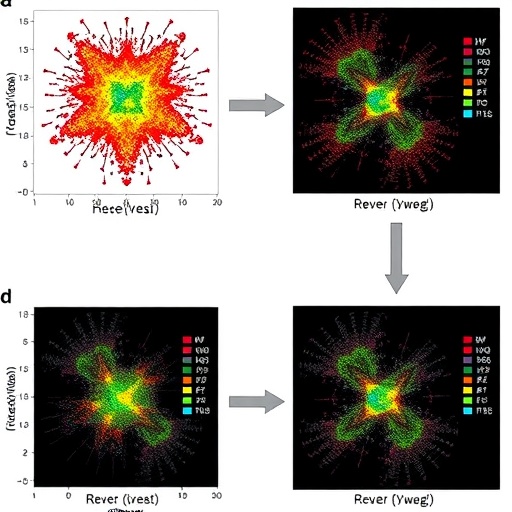
Enter Fountain, a pioneering deep learning framework designed for the rigorous integration of scATAC-seq datasets utilizing a novel approach known as regularized barycentric mapping. This innovative methodology leverages the principles of optimal transport theory to facilitate the transformation of one data distribution into another in a mathematically sound manner. The incorporation of geometric data information into the barycentric mapping process acts as a regularization factor, ensuring that the biological variance present in the original distributions is preserved during integration.
One of the remarkable features of Fountain is its ability to achieve accurate batch alignment without compromising the diversity inherent in biological samples. This advantage was demonstrated through comprehensive experimental validations across a myriad of real-world datasets, where Fountain consistently outperformed existing integration methods. The results underscored Fountain’s capability not just to correct for batch artifacts effectively but also to uphold the biological nuances that exist among different cell types.
Moreover, a standout characteristic of Fountain is its adaptability to the integration of new batches alongside already processed data without requiring a full retraining of the model. This continuous online capacity signifies a leap forward in the integration processes for scATAC-seq datasets, allowing researchers to include new samples as they become available while maintaining consistency with previously analyzed data. This feature is paramount in rapidly evolving research environments, where the dynamic accumulation of data necessitates a flexible and efficient integration solution.
Beyond integration, Fountain’s reconstruction strategy holds immense potential in generating batch-corrected ATAC profiles. This capability not only enhances the fidelity with which cellular heterogeneity is captured but also facilitates deeper insights into cell-type-specific functions. For instance, researchers can perform expression enrichment analyses to identify genes that are differentially accessed among distinct cell populations, revealing critical biological insights regarding cellular functions, lineage differentiation, and disease states.
As the need for accurate genomic data integration grows, particularly in studies involving large and complex datasets from diverse biological contexts, Fountain emerges as a crucial tool that meets these demands. Its innovative approach to seamless integration, combined with its ability to preserve essential biological features, marks a significant advancement in the computational biology toolkit available to researchers today.
The implications of the Fountain framework extend far beyond batch correction alone. It sets a new standard for how researchers can approach the integration of single-cell genomic data. By seamlessly aligning datasets from various sources without losing biological context, Fountain empowers scientists to conduct more reliable and insightful analyses. This methodology embodies a critical evolution in how we engage with high-dimensional genomic data, enabling the extraction of insights that were previously obscured by technical artifacts.
Ultimately, the introduction of Fountain into the realm of scATAC-seq data integration exemplifies the synergy between advanced computational techniques and biological research. As a result, researchers can unlock additional layers of genomic information, leading to a better understanding of gene regulation and cellular behavior in health and disease.
The rapid pace of progress in sequencing technologies, coupled with innovative integration tools like Fountain, heralds a new era in genomic exploration. It promises not only to enhance the reliability of data interpretation but also to push the boundaries of what scientists can achieve when interpreting the intricate landscapes of chromatin accessibility. With such tools, the scientific community is better equipped to tackle the complexities of gene regulation, unveiling the secrets of the genome and providing insight into the biological phenomena that shape life.
As we stand on the brink of this genomic revolution, it is clear that tools like Fountain will play a pivotal role in the collective effort to decode the complexities of biology. By ensuring that our analyses remain as true to the biological reality as possible, we can aspire to uncover new therapeutic strategies, enhance our understanding of genetic diseases, and ultimately, inform precision medicine approaches tailored to the unique genetic makeup of individual patients.
In summary, the integration of scATAC-seq data presents significant challenges owing to batch effects that can obscure biological variations. However, the innovative Fountain framework, with its approach grounded in rigorous barycentric mapping, offers a robust solution to these challenges. By enabling the preservation of biological heterogeneity while correcting for batch-related discrepancies, Fountain represents a leap forward in our ability to analyze complex genomic data. As we move forward in this era of genomics, tools like Fountain will undoubtedly shape the landscape of biological research.
Subject of Research: Integration of single-cell ATAC-seq data
Article Title: Rigorous integration of single-cell ATAC-seq data using regularized barycentric mapping
Article References: Zhu, S., Hua, H. & Chen, S. Rigorous integration of single-cell ATAC-seq data using regularized barycentric mapping. Nat Mach Intell 7, 1461–1477 (2025). https://doi.org/10.1038/s42256-025-01099-3
Image Credits: AI Generated
DOI: https://doi.org/10.1038/s42256-025-01099-3
Keywords: scATAC-seq, batch effects, data integration, genomic data analysis, computational biology, deep learning, barycentric mapping, gene regulation, chromatin accessibility, optimal transport.
Tags: batch effect correction methodsbiological dataset integrationchromatin accessibility analysisdata harmonization toolsgene regulation dynamicsgenomic research innovationsgenomics data integrationregulatory landscape explorationscATAC-seq challengessingle-cell ATAC-seq techniquessingle-cell sequencing methodssingle-cell technologies advancement


{kind=link}