Credit: KAIST
A deep learning-powered computational framework, ‘DeepEC,’ will allow the high-quality and high-throughput prediction of enzyme commission numbers, which is essential for the accurate understanding of enzyme functions.
A team of Dr. Jae Yong Ryu, Professor Hyun Uk Kim, and Distinguished Professor Sang Yup Lee at KAIST reported the computational framework powered by deep learning that predicts enzyme commission (EC) numbers with high precision in a high-throughput manner.
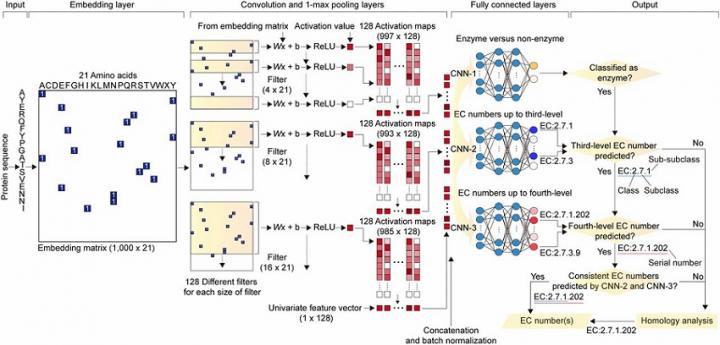
DeepEC takes a protein sequence as an input and accurately predicts EC numbers as an output. Enzymes are proteins that catalyze biochemical reactions and EC numbers consisting of four level numbers (i.e., a.b.c.d) indicate biochemical reactions. Thus, the identification of EC numbers is critical for accurately understanding enzyme functions and metabolism.
EC numbers are usually given to a protein sequence encoding an enzyme during a genome annotation procedure. Because of the importance of EC numbers, several EC number prediction tools have been developed, but they have room for further improvement with respect to computation time, precision, coverage, and the total size of the files needed for the EC number prediction.
DeepEC uses three convolutional neural networks (CNNs) as a major engine for the prediction of EC numbers, and also implements homology analysis for EC numbers if the three CNNs do not produce reliable EC numbers for a given protein sequence. DeepEC was developed by using a gold standard dataset covering 1,388,606 protein sequences and 4,669 EC numbers.
In particular, benchmarking studies of DeepEC and five other representative EC number prediction tools showed that DeepEC made the most precise and fastest predictions for EC numbers. DeepEC also required the smallest disk space for implementation, which makes it an ideal third-party software component.
Furthermore, DeepEC was the most sensitive in detecting enzymatic function loss as a result of mutations in domains/binding site residue of protein sequences; in this comparative analysis, all the domains or binding site residue were substituted with L-alanine residue in order to remove the protein function, which is known as the L-alanine scanning method.
This study was published online in the Proceedings of the National Academy of Sciences of the United States of America (PNAS) on June 20, 2019, entitled “Deep learning enables high-quality and high-throughput prediction of enzyme commission numbers.”
“DeepEC can be used as an independent tool and also as a third-party software component in combination with other computational platforms that examine metabolic reactions. DeepEC is freely available online,” said Professor Kim.
Distinguished Professor Lee said, “With DeepEC, it has become possible to process ever-increasing volumes of protein sequence data more efficiently and more accurately.”
###
This work was supported by the Technology Development Program to Solve Climate Changes on Systems Metabolic Engineering for Biorefineries from the Ministry of Science and ICT through the National Research Foundation of Korea. This work was also funded by the Bio & Medical Technology Development Program of the National Research Foundation of Korea funded by the Korean government, the Ministry of Science and ICT.
Media Contact
Younghye Cho
[email protected]
Original Source
https:/
Related Journal Article
http://dx.


{kind=link}