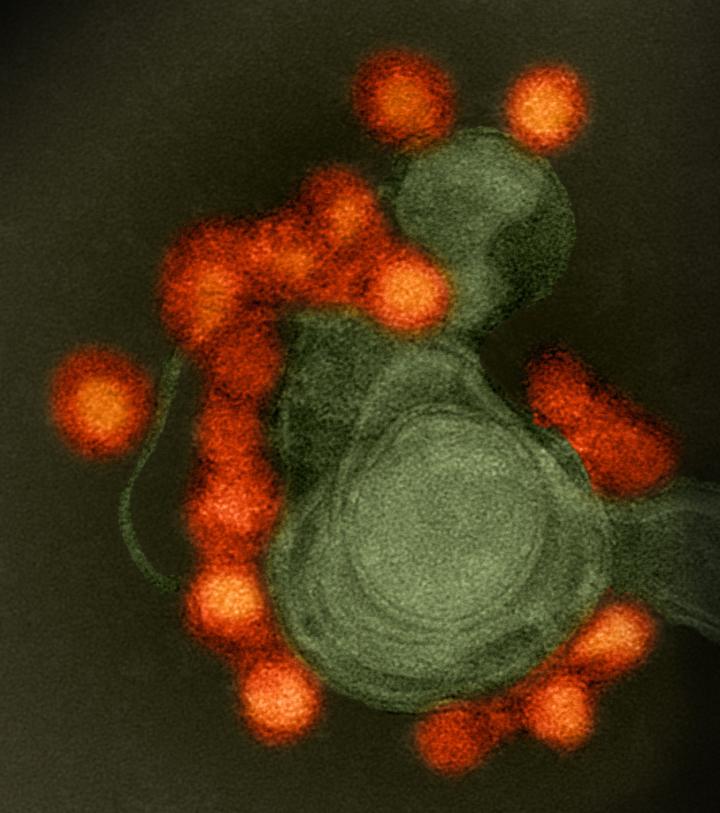
Credit: NIAID
WHAT:
Genetic analysis of samples collected as the Zika virus (ZIKV) spread throughout the Americas after its introduction in 2013 or 2014 has shown that the virus circulated undetected for up to a year in some regions before it came to the attention of public health authorities. Genetic sequencing has also enabled scientists to recreate the epidemiological and evolutionary paths the virus took as it spread and split into the distinct subtypes–or clades–that have been detected in the Americas. The research, published in Nature today, was supported in part by the National Institute of Allergy and Infectious Diseases (NIAID), part of the National Institutes of Health.
The researchers reconstructed Zika's dispersal by sequencing ZIKV genetic material collected from hundreds of patients in ten countries and territories. They eventually amassed a database of 110 complete or partial ZIKV genomes–the largest collection to date–which they analyzed along with 64 published and publicly shared genomes.
Based on changes to the viral genome that accumulated as the disease moved through new populations, the researchers concluded that ZIKV spread rapidly upon its initial introduction in Brazil, likely sometime in 2013. Later, at several points in early-to-mid 2015, the virus separated into at least three clades–distinct genetic groups whose members share a common ancestor–in Colombia, Honduras, and Puerto Rico, as well as a fourth type found in parts of the Caribbean and the continental United States.
The researchers used innovative sequencing approaches to overcome the challenge of working with samples not originally intended for viral genetic sequencing. In future outbreaks, the researchers suggest, appropriate samples for genetic sequencing should be collected from the beginning to make retrospective and concurrent analysis much easier and more accurate.
These results also may have a direct impact on public health, as a clear understanding of the genetic changes to pathogens that can occur during emerging outbreaks will be key to informing disease surveillance and development of diagnostic tests. Similarly, genetic analysis and prompt sharing in public databases can enhance the understanding of the dynamics of disease spread, so that public health authorities may implement the best possible control and prevention strategies.
###
ARTICLE:
HC Metsky et al. Genome sequencing reveals Zika virus diversity and spread in the Americas. Nature DOI: 10.1038/nature22402 (2017).
WHO:
NIAID Director Anthony S. Fauci, M.D., is available to comment. Liliana L. Brown., Ph.D., a program officer in the Office of Genomics and Advanced Technologies in NIAID's Division of Microbiology and Infectious Diseases (DMID), and Cristina Cassetti, Ph.D., chief of the Virology Branch in DMID, are also available for comment.
CONTACT:
To schedule interviews, please contact Elizabeth Deatrick, (301) 402-1663, [email protected]
NIAID conducts and supports research–at NIH, throughout the United States, and worldwide–to study the causes of infectious and immune-mediated diseases, and to develop better means of preventing, diagnosing and treating these illnesses. News releases, fact sheets and other NIAID-related materials are available on the NIAID website.
About the National Institutes of Health (NIH): NIH, the nation's medical research agency, includes 27 Institutes and Centers and is a component of the U.S. Department of Health and Human Services. NIH is the primary federal agency conducting and supporting basic, clinical, and translational medical research, and is investigating the causes, treatments, and cures for both common and rare diseases. For more information about NIH and its programs, visit http://www.nih.gov.
NIH…Turning Discovery Into Health®
Media Contact
Elizabeth Deatrick
[email protected]
301-402-1663
@NIAIDNews
http://www.niaid.nih.gov
Related Journal Article
http://dx.doi.org/10.1038/nature22402

{kind=link}