Credit: (C) InSysBio
Moscow, Russia, September 28, 2017 – One must activate amyloid degradation as soon as possible to prevent the appearance of the protein plaques in brain under Alzheimer's conditions. This conclusion was reached by scientists from the biotechnology company InSysBio, who created the first computer model of the disease. The final part of the work has been recently published in CPT: Pharmacometrics & Systems Pharmacology.
Alzheimer's disease is an age-related disease that leads to a serious cognitive impairment. Traditionally, investigators associate the development of the pathology with accumulation of the beta amyloid protein. Normally, it is an important component of the neuron regulation process, but in large concentrations it becomes toxic, causes inflammatory processes in brain and aggregates into insoluble amyloid plaques. Pharmacologists who are searching for an effective treatment consider that if one stops the accumulation of this protein and its aggregation into plaques, the disease can be cured. However, it is difficult to predict how modern therapies can affect the progression of amyloid pathology, how it depends on the stage of the disease and how to prevent the accumulation of protein without violating its natural functions.
A group of scientists from the R&D company InSysBio investigates this problem with mathematical accuracy – they created a computer model of amyloid beta accumulation.
Intensive development took them several years. Scientists took information from published papers and clinical studies, analyzed it and described the laws by equations. Although there is a huge amount of the data on this topic in public access, no one has yet systematized and analyzed them. The pioneers were Tatiana Karelina's group of modeling neurodegenerative diseases.
'We have already published the first part of our study. Previous model described the amount of amyloid in the normal or pathological states, but not the transition between them, so it was not possible to investigate the progression of the disease. The results of studies for amyloid plaques were added to consider the whole process', Tatiana Karelina explains.
First of all, scientists focused on the data of the direct post mortem biochemical brain studies of the patients who died from the disease at different age. In addition, they included the analysis of the results of clinical trials of anti-Alzheimer's therapies, most of which failed due to insufficient effectiveness. The desire to help pharmacologists understand the problem was the main motivation for the team.
The model was designed in such a way so that the simulations can be translated into the results of traditional diagnostic methods for Alzheimer's – positron emission tomography (PET) and measuring the concentration of amyloid in the cerebrospinal fluid. These tests are commonly performed to diagnose a disease or determine a risk group. InSyBio scientists used them to check the accuracy and calibrate the entire model.
As the result, the group derived a system of equations with 30 variables describing the main processes that lead to the accumulation of amyloid in Alzheimer's disease, which are
- Increased protein production in the brain
- Decreased degradation (destruction) of amyloid
- Activation of aggregation of amyloid protein
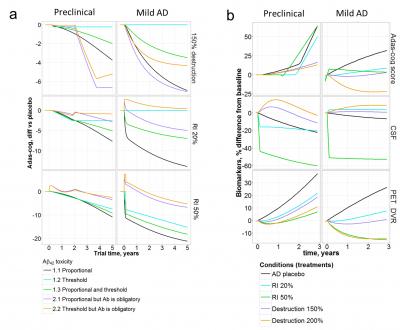
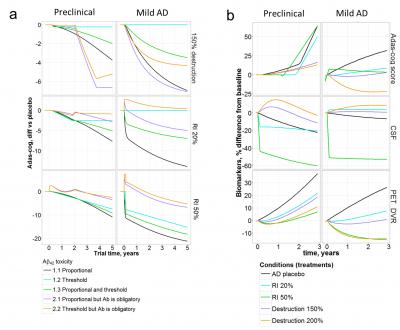
Varying the parameters of contribution of each of the listed processes to the development of amyloid pathology, Tatiana and colleagues found that the main cause was the violation of the insoluble protein form degradation.
Scientists managed to connect three important markers of the Alzheimer's disease: the content of soluble amyloid in the brain, in the cerebrospinal fluid and the amount of insoluble plaques.
But most of all the researchers were interested whether modern drugs could lead the main markers of the amyloid pathology to the normal physiological values. After simulating the long-term usage of modern therapies at different stages of the disease, the scientists found that only activation of beta-amyloid degradation leads to a relative normalization of the indices. At the same time, in order to achieve a positive result, it was necessary to start taking the drug at the earliest stages of the pathology development – at the age of 60 years.
For the final testing of the model, the team has chosen a complex task: the researchers have checked whether the program could predict improvements in patients cognitive functions.
Firstly, Tatiana's group formulated four of the most popular hypotheses about the toxicity of amyloid, which drove several recent clinical trials. That is, researchers have linked different forms of amyloid with damage to brain cells and cognitive loss which is estimated in clinical practice by the Adas-cog test. The hypotheses were tested by including appropriate treatment into the model. For example, to assess the hypothesis of the amyloid soluble form toxicity, the researchers simulated the effect of the real drug, suppressing protein synthesis. It turned out that many of the hypotheses could be verified by the data of clinical trials and predictions of a new computer model. And this means that future clinical trials can be planned more effectively.
'We don't make loud statements that we have understood the cause of Alzheimer's disease. We show that the model can assist to estimate the hypotheses quickly and effectively. The most important question to answer was how to help the patient? With our model we managed to determine the optimal treatment scheme to normalize the concentration of all amyloid forms, but it still remains unknown whether this will affect cognitive functions and improve the patient's life. This requires real clinical trials', says Tatiana.
Now the team is preparing the model for the presentation at the American Conference on Pharmacometrics – the most important annual event for innovative drug developers. Scientists hope not only to discuss their results with the community, but also to find new partners for further work.
###
Media Contact
Maria Maximova
[email protected]
@insysbio
http://insysbio.ru/en
Related Journal Article
http://dx.doi.org/10.1002/psp4.12249

{kind=link}