In a groundbreaking study poised to redefine therapeutic strategies for muscular dystrophies, a team of researchers led by Fu, Wang, and Zhang have unveiled compelling evidence that mutations in the GMPPB gene precipitate dystroglycanopathy symptoms in mice, which can be effectively reversed through targeted molecular interventions. Published in Nature Communications in 2026, this research not only sheds light on the elusive pathological mechanisms underlying dystroglycanopathies but also demonstrates the remarkable potential of GSK3β inhibition and adeno-associated virus (AAV)-mediated gene therapy as viable treatments.
Dystroglycanopathies represent a subset of congenital muscular dystrophies characterized by defective glycosylation of α-dystroglycan, a critical protein that anchors the extracellular matrix to the cytoskeleton in muscle fibers and neural tissues. Mutations in various genes encoding glycosyltransferases disrupt this process, leading to a spectrum of clinical symptoms ranging from muscle weakness to severe brain malformations. Among these, mutations in the gene encoding GDP-mannose pyrophosphorylase B (GMPPB) have remained relatively underexplored, despite their clinical significance in human patients presenting with limb-girdle muscular dystrophy and congenital myasthenic syndromes.
The research team engineered a mouse model harboring targeted mutations within the Gmppb locus, which recapitulated key features of human dystroglycanopathy. These mice exhibited severe motor deficits, reduced muscle mass, and aberrant glycosylation patterns of α-dystroglycan, confirming the pathogenic role of Gmppb mutations. Importantly, histological analyses revealed extensive muscle fiber necrosis and fibrosis, accompanied by a compromised basal lamina structure, hallmarking the cascade of degenerative events triggered by defective GMPPB function.
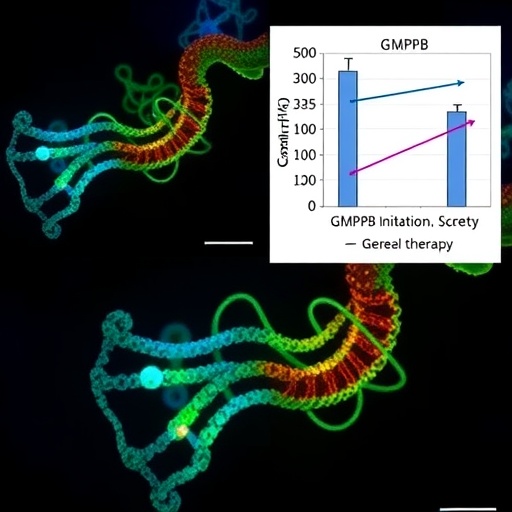
Crucially, the study delved into the signaling pathways perturbed by GMPPB insufficiency. Glycogen synthase kinase 3 beta (GSK3β), a serine/threonine kinase implicated in diverse cellular processes including glycogen metabolism and protein synthesis, emerged as a pivotal modulator of α-dystroglycan glycosylation. Pharmacological inhibitors targeting GSK3β demonstrated robust efficacy in restoring glycosylation profiles and ameliorating muscle pathology in the mutant mice. This therapeutic effect suggests that aberrant activation of GSK3β contributes to the dystroglycanopathy phenotype and that its modulation offers a promising avenue for intervention.
The second arm of the study explored gene replacement strategies employing AAV vectors to deliver functional GMPPB copies directly into affected tissues. The vector demonstrated high tropism for skeletal muscle and central nervous system cells, enabling efficient transgene expression without eliciting significant immunogenicity. Treated mutant mice exhibited marked improvements in motor function, muscle histopathology, and lifespan extension, underscoring the transformative potential of gene therapy for inherited muscular dystrophies.
Beyond skeletal muscle, the research illuminated the systemic impact of GMPPB mutations. Neurodevelopmental assessments revealed cortical malformations and defective neuronal migration in mutant animals, consistent with human clinical presentations of dystroglycanopathies. Intriguingly, AAV-mediated gene delivery also ameliorated central nervous system abnormalities, indicating that therapeutic targeting of GMPPB can transcend peripheral tissues to address multisystemic pathology.
Mechanistically, the authors propose that GMPPB mutations disrupt the biosynthesis of GDP-mannose, a critical sugar nucleotide donor for O-mannosylation pathways responsible for post-translational modification of α-dystroglycan. This deficit compromises the assembly of functional glycan moieties, thereby impairing the dystroglycan extracellular matrix receptor complex. By restoring GDP-mannose availability through gene replacement or attenuating downstream signaling aberrations via GSK3β inhibition, cellular homeostasis and extracellular matrix integrity are reinstated.
The implications of these findings resonate far beyond the immediate scope of dystroglycanopathies. They exemplify the convergence of gene editing, molecular pharmacology, and regenerative medicine, forging a path toward precision treatments tailored to genetic underpinnings. Moreover, the dual therapeutic approach underscores the necessity of multifaceted strategies when confronting complex glycosylation disorders, which often involve intricate networks of enzymatic cascades and signaling pathways.
This pioneering work gains significance amidst the broader context of muscular dystrophy research where effective treatments remain scarce. By elucidating the role of GMPPB in muscle and neural pathology and demonstrating tangible rescue mechanisms, the study invigorates ongoing efforts to develop gene-based cures for these debilitating diseases. Furthermore, it sets a benchmark for future research aimed at deciphering the molecular etiologies and correcting glycosylation defects in inherited disorders.
The versatility of AAV vectors in delivering therapeutic genes with high specificity and long-term expression catalyzes optimism for clinical translation. Coupled with the availability of small-molecule inhibitors like those targeting GSK3β, the research presents an immediate platform for combinatorial therapies that could be rapidly advanced into clinical trials. The prospect of reversible interventions for progressive muscular dystrophies holds profound promise for patients and their families, potentially altering disease trajectories and improving quality of life.
Importantly, the comprehensive phenotyping of the Gmppb-mutant mouse model enriches the genetic toolkit available for dystroglycanopathy research, facilitating the exploration of genotype-phenotype correlations and the identification of modifier genes. Such models are indispensable for preclinical testing, enabling the optimization of dosing regimens, delivery methods, and combinational strategies to maximize therapeutic benefit while minimizing side effects.
The elucidation of GSK3β’s role in dystroglycanopathies further invites investigation into its broader involvement in other glycosylation disorders and neuromuscular diseases. As a central node in multiple signaling cascades, GSK3β presents a tantalizing target whose modulation may yield dividends in diverse pathological contexts. However, the challenge remains to achieve selective inhibition with minimal off-target effects, necessitating continued medicinal chemistry efforts.
Moreover, the study highlights the importance of temporal intervention windows. Early postnatal delivery of AAV-GMPPB yielded superior outcomes, emphasizing the need for prompt diagnosis and therapeutic initiation in clinical settings. This insight advocates for enhanced newborn screening programs and genetic counseling to identify at-risk individuals before irreversible tissue damage ensues.
In synthesizing these complex findings, Fu, Wang, and Zhang not only advance the molecular understanding of dystroglycanopathies but also pioneer translational strategies that could soon transition from bench to bedside. Their comprehensive approach exemplifies the future of neuromuscular therapeutics, where genetic precision and molecular innovation converge to surmount previously intractable diseases.
As the medical community grapples with the challenges posed by congenital myopathies, studies like this illuminate a hopeful path forward. The integration of gene replacement therapies with targeted pathway inhibition represents a powerful paradigm shift, heralding a new era of personalized medicine tailored to the intricacies of individual genetic mutations. Patients afflicted with severe dystroglycanopathies may soon witness tangible improvements attributable to these scientific breakthroughs.
In conclusion, the demonstration that Gmppb mutations induce dystroglycanopathy phenotypes in mice, which can be rescued by GSK3β inhibition or AAV-mediated GMPPB gene replacement, marks a pivotal advance in muscular dystrophy research. This multifaceted therapeutic approach establishes a framework for future interventions, promising to redefine clinical management and improve outcomes for patients suffering from these debilitating disorders. The broader implications for glycosylation biology and gene therapy further amplify the study’s impact, charting an ambitious course for next-generation neuromuscular disease treatment.
Subject of Research: The study investigates the pathological role of GMPPB mutations in dystroglycanopathies and evaluates therapeutic interventions using GSK3β inhibition and AAV-mediated GMPPB gene replacement.
Article Title: Gmppb-mutant mice exhibit dystroglycanopathy symptoms that are rescued with GSK3β inhibition or AAV-mediated GMPPB gene replacement.
Article References:
Fu, Z., Wang, T., Zhang, C. et al. Gmppb-mutant mice exhibit dystroglycanopathy symptoms that are rescued with GSK3β inhibition or AAV-mediated GMPPB gene replacement. Nat Commun (2026). https://doi.org/10.1038/s41467-026-71524-7
Image Credits: AI Generated
Tags: AAV-mediated gene therapy muscular dystrophycongenital muscular dystrophy treatmentdystroglycanopathy molecular mechanismsgene therapy for dystroglycanopathiesglycosyltransferase mutation effectsGMPPB gene mutation muscular dystrophyGMPPB mutation mouse modelGSK3β inhibition therapylimb-girdle muscular dystrophy animal modelmuscle weakness genetic causestargeted molecular interventions muscular disordersα-dystroglycan glycosylation defects

{kind=link}