
In a groundbreaking study set to redefine our understanding of autoimmune blistering diseases, researchers have uncovered a pivotal mechanism by which the immune system’s own antibodies incite a damaging inflammatory cascade in skin cells. The study, published in Nature Communications, delves into the intricate molecular dialogue between IgG autoantibodies and keratinocytes, the predominant cells of the epidermis, revealing a critical involvement of MyD88-dependent pro-inflammatory signaling. This discovery not only illuminates the pathogenic underpinnings of bullous pemphigoid (BP) but also opens promising therapeutic avenues that could revolutionize treatment for millions suffering from this debilitating skin disorder.
Bullous pemphigoid is an autoimmune condition characterized by the formation of large, fluid-filled blisters predominantly affecting elderly populations. The disease arises when the body erroneously generates IgG autoantibodies against components of the dermal-epidermal junction, triggering skin detachment. Until now, the exact cellular and molecular consequences of these autoantibodies interacting with keratinocytes remained poorly understood. The team, led by Bao and colleagues, employed a combination of molecular biology techniques, patient-derived samples, and sophisticated in vitro models to probe this interaction in unprecedented detail.
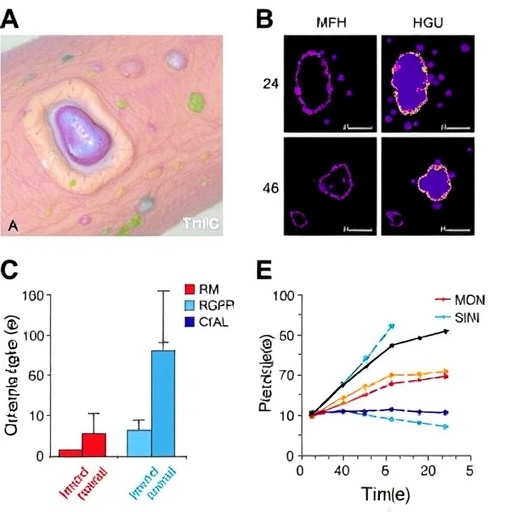
Central to their findings is the role of MyD88, an adaptor protein integral to Toll-like receptor (TLR) signaling pathways known for mediating innate immune responses. The researchers demonstrated that binding of IgG autoantibodies to keratinocytes initiates a MyD88-dependent pro-inflammatory program within these cells. This signaling cascade culminates in the production of cytokines and chemokines, molecules that perpetuate local inflammation and recruit immune cells, ultimately exacerbating tissue damage and blister formation. This mechanistic insight challenges the traditional view of keratinocytes as passive victims in BP and instead positions them as active participants driving disease pathology.
.adsslot_MwRUEmk7yo{width:728px !important;height:90px !important;}
@media(max-width:1199px){ .adsslot_MwRUEmk7yo{width:468px !important;height:60px !important;}
}
@media(max-width:767px){ .adsslot_MwRUEmk7yo{width:320px !important;height:50px !important;}
}
ADVERTISEMENT
The study utilized keratinocyte cultures exposed to patient-derived IgG autoantibodies and employed gene silencing strategies to abrogate MyD88 expression. Remarkably, the suppression of MyD88 significantly attenuated the inflammatory response, evidencing the essential role of this adaptor protein. Furthermore, analyses of skin biopsies from BP patients supported these in vitro findings, revealing upregulated expression of MyD88 and downstream inflammatory mediators within lesional epidermis. These human data lend strong translational relevance to their mechanistic conclusions.
Intriguingly, this MyD88-dependent pathway appears distinct from classical antibody-mediated cytotoxicity, suggesting that autoantibody engagement triggers a non-canonical signaling axis within keratinocytes. This paradigm-shifting observation expands the conceptual framework of autoimmune blistering diseases beyond immune effector cell infiltration and complement activation, highlighting keratinocyte-intrinsic signaling as a critical driver of inflammation. Such insights carry profound implications for the design of targeted therapies that could interrupt this pathogenic feedback loop at its source.
Notably, the pro-inflammatory signature orchestrated by MyD88 within keratinocytes encompasses multiple cytokines, including interleukin-6 (IL-6), tumor necrosis factor-alpha (TNF-α), and various chemokines. These mediators contribute to the recruitment and activation of circulating immune cells, amplifying the local inflammatory milieu. Continuous secretion of these factors by keratinocytes may sustain chronic inflammation, explaining the persistence and progression of lesions in BP patients. Targeting these signaling nodes could alleviate symptoms and hinder disease advancement.
From a therapeutic standpoint, inhibitors of MyD88 or its downstream signaling molecules represent attractive candidates for drug development. Currently, treatments for BP primarily rely on systemic immunosuppression, which carries significant side effects and risks in elderly populations. The identification of keratinocyte MyD88 as a key modulator introduces the possibility of more selective interventions that mitigate inflammation without broadly dampening immune function. Such precision medicine approaches could markedly improve patient quality of life and outcomes.
Moreover, this study lays the groundwork for exploring MyD88-dependent responses in other autoimmune and inflammatory skin diseases. Given the ubiquity of TLRs and their adaptors in immune signaling, similar pathways may contribute to pathology in conditions such as pemphigus vulgaris or lupus erythematosus. Systematic investigation across disease contexts may uncover shared mechanisms that underpin skin inflammation and barrier disruption, enabling cross-cutting therapeutic strategies.
The researchers also observed that the magnitude of the MyD88-mediated response correlates with disease severity, suggesting potential utility as a biomarker for clinical prognosis. Quantifying expression levels of MyD88 or related inflammatory molecules in patient skin samples could inform individualized treatment regimens. Early identification of aggressive disease phenotypes would allow prompt intervention, potentially curbing lesion formation before extensive tissue damage occurs.
This work exemplifies the power of integrating patient-derived biological materials with cutting-edge molecular techniques. By bridging bench and bedside, Bao et al. advance both fundamental immunology and translational medicine. Their methodology includes high-resolution immunostaining, gene knockout systems, and transcriptomic profiling, which together yield robust, multilayered data validating the MyD88-dependent inflammatory axis in BP keratinocytes.
While the study profoundly enhances current understanding, it also invites further questions. How do IgG autoantibodies mechanistically engage receptors on keratinocytes to initiate MyD88 signaling? Are other adaptor proteins or co-receptors involved in modulating these responses? What are the long-term effects of inhibiting this pathway in vivo, particularly on skin homeostasis and immunity? Addressing these queries will be critical toward harnessing this knowledge for clinical benefit.
Furthermore, the interplay between keratinocyte-intrinsic pathways and the broader immune system warrants deeper exploration. The cross-talk between epithelial cells, resident immune cells, and infiltrating leukocytes likely orchestrates the complex tissue landscape observed in BP lesions. Dissecting these cellular interactions at single-cell resolution could unravel novel pathogenic circuits amenable to intervention.
In summary, the discovery of an IgG autoantibody-induced, MyD88-dependent pro-inflammatory response in keratinocytes constitutes a major leap forward in understanding bullous pemphigoid pathogenesis. By repositioning keratinocytes as active drivers rather than mere targets of inflammation, this research revolutionizes conceptual paradigms and highlights new molecular targets for therapy. Its implications extend beyond one disease, potentially reshaping approaches to a spectrum of autoimmune skin disorders.
As science advances toward an era of personalized medicine, insights such as these exemplify the critical importance of elucidating cellular signaling nuances underlying chronic inflammatory diseases. The findings reported by Bao and colleagues provide a beacon for future endeavors aimed at developing safer, more effective treatments that improve patient outcomes and minimize collateral damage from immune dysregulation.
This landmark study is poised to generate widespread excitement in dermatological research and clinical practice, catalyzing innovative strategies that transcend conventional immunosuppressive therapies. Ultimately, it reinforces the profound impact of molecular immunology in decoding the complexities of human disease and charting paths toward cures.
Subject of Research: The molecular mechanisms by which IgG autoantibodies in bullous pemphigoid induce pro-inflammatory responses in keratinocytes via a MyD88-dependent pathway.
Article Title: IgG autoantibodies in bullous pemphigoid induce a pathogenic MyD88-dependent pro-inflammatory response in keratinocytes.
Article References:
Bao, L., Guerrero-Juarez, C.F., Li, J. et al. IgG autoantibodies in bullous pemphigoid induce a pathogenic MyD88-dependent pro-inflammatory response in keratinocytes. Nat Commun 16, 7254 (2025). https://doi.org/10.1038/s41467-025-62495-2
Image Credits: AI Generated
Tags: autoimmune blistering diseasesbullous pemphigoid mechanismsepidermal immune responseIgG autoantibodieskeratinocytes and skin healthmolecular biology in autoimmune researchMyD88-driven inflammationpatient-derived samples in studiespro-inflammatory signaling pathwaysskin detachment and blisterstherapeutic strategies for skin disordersToll-like receptor signaling


{kind=link}