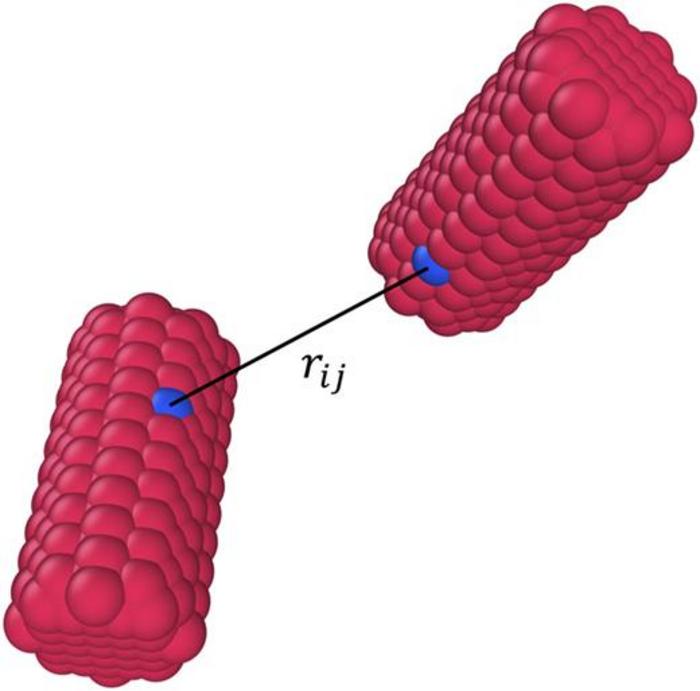
Simulating particles is a relatively simple task when those particles are spherical. In the real world, however, most particles are not perfect spheres but take on irregular and varying shapes and sizes. Simulating these particles becomes a much more challenging and time-consuming task.
Simulating particles is a relatively simple task when those particles are spherical. In the real world, however, most particles are not perfect spheres but take on irregular and varying shapes and sizes. Simulating these particles becomes a much more challenging and time-consuming task.
The ability to simulate particles is critical to understanding how they behave. For example, microplastics are a new form of pollution as plastic waste has increased drastically and uncontrollably decays in the environment by either mechanical means or UV degradation. These very tiny particles are now found nearly everywhere in the world. To be able to remedy this environmental crisis, it is important to understand more about these particles and how they behave.
In an effort to combat this challenge, researchers at the University of Illinois Urbana-Champaign have trained neural networks to predict interactions between irregularly shaped particles to accelerate molecular dynamics simulations. With this method, simulations can be done up to 23 times faster compared to traditional simulation methods and can be applied to any irregular shape with sufficient training data.
“Microplastics are now present everywhere in the environment and most of them are not spheres, they are very heterogeneous, and they have corners and edges. Tackling the problem of how they behave in the environment requires us to develop new methods, finding ways to simulate them faster, cheaper and more efficiently,” says Antonia Statt, professor of materials science and engineering.
Spheres are easy to simulate because the only parameter needed to determine how two particles interact is the distance between the centers each sphere. Moving from a sphere to more complicated shapes—like cubes or cylinders—requires knowing not only how far away two particles are from one another, but also the angles and the relative positions of each particle. The traditional method of simulating cubes, for example, involves building the cube out of many little spheres.
“It’s a very roundabout way of describing a cube, to tessellate it with small spheres,” Statt explains. “It’s also expensive because you have to calculate the interactions of all the little spheres with each other. To bypass that, we used machine learning—a feed forward neural net—which is a fancy way of saying, ‘let’s fit a complicated function that we don’t know.’ And neural nets are really good at that. If you provide them with enough data, they can fit anything you like.”
Using this method, all the distances between the little spheres don’t need to be calculated individually. Only the cube center-to-center distance and its relative orientation is needed, making it much easier and faster. Further, this method is as accurate as traditional methods. It cannot be more accurate since it is trained on data produced from traditional methods, but it can be more efficient.
In the future, Statt would like to be able to simulate more complicated irregular shapes as well as mixtures of different shapes, like a cube and a cylinder rather than two cubes. “We will have to learn all the individual interactions, but the method is general enough that we will be able to do that,” she says.
This research, “Molecular dynamics simulations of anisotropic particles accelerated by neural-net predicted interactions,” was recently published in The Journal of Chemical Physics and was selected for the 2024 JCP Emerging Investigators Special Collection. It also features on the cover of this issue of JCP.
Antonia Statt is also an affiliate of the Materials Research Laboratory, the department of chemical and biomolecular engineering and the Beckman Institute for Advanced Science and Technology at Illinois.
Other contributors to this work include B. Ruşen Argun (department of mechanical engineering, Illinois) and Yu Fu (department of physics, Illinois).
This research was funded by the Molecule Maker Lab Institute (MMLI): An AI Research Institutes program supported by the National Science Foundation.
Journal
The Journal of Chemical Physics
DOI
10.1063/5.0206636
Article Title
Molecular dynamics simulations of anisotropic particles accelerated by neural-net predicted interactions
Article Publication Date
21-Aug-2024

{kind=link}