All cells in an organism contain identical DNA sequence. What determines the identity and function of individual cells and tissues, is the set of genes that will be active in a given place, at a given time. These active genes are transcribed from the DNA template into distinct messenger RNA (mRNA) molecules and will encode the proteins the cell needs to function.
Credit: MPI of Immunobiology & Epigenetics, Hilgers
All cells in an organism contain identical DNA sequence. What determines the identity and function of individual cells and tissues, is the set of genes that will be active in a given place, at a given time. These active genes are transcribed from the DNA template into distinct messenger RNA (mRNA) molecules and will encode the proteins the cell needs to function.
At specific places called promoters, a complex molecular machinery starts transcribing DNA sequences into mRNA. Interestingly, most genes contain multiple possible sites where transcription can start or end. This means that for each gene, depending on the start or termination site, the mRNAs can be different. Expressing one gene in different variants expands the diversity and functionality of the genome many times over. At the same time, it adds another layer of complexity to the study the genome.
RNA snapshots from beginning to end
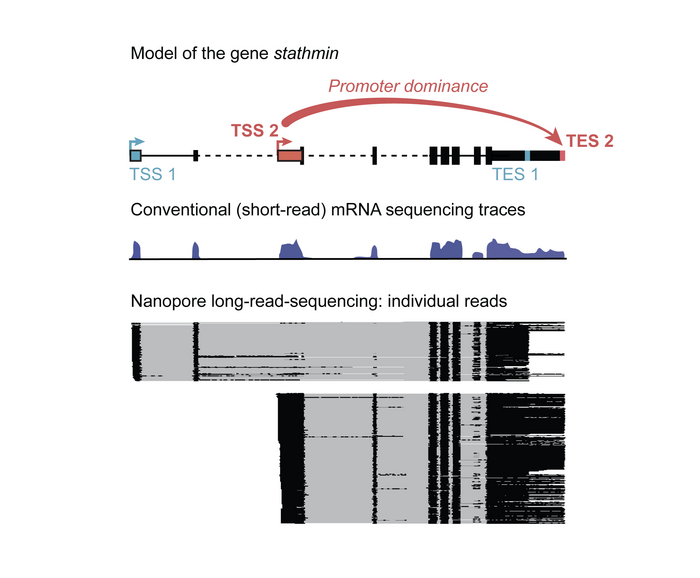
Scientists at the Max Planck Institute of Immunobiology and Epigenetics in Freiburg wanted to know how many different start and end sites each gene uses, in which combination, and whether the combinations were different in different conditions. “The technical problem to answer this question is that we have to “read” each and every mRNA molecule from all genes from the very beginning to the very end. This a humongous task that has not been undertaken before,” says Valérie Hilgers, a research group leader at the MPI-IE.
The scientists used a tweaked next-generation sequencing technology to read out the individual mRNAs. For conventional short-read sequencing, each mRNA is broken into shorter fragments that are amplified and then sequenced to produce the “read”. Bioinformatic techniques are then used to piece together the reads like a jigsaw, into a continuous sequence. For full-length mRNA information of the entire genome in several Drosophila tissues, including the brain, the Hilgers teamed up with the Deep Sequencing Facility of the MPI to optimize specific long-read-sequencing technologies. “Long-read sequencing allows for the retrieval of much longer sequencing reads than widely used standard sequencing. However, we even had to optimize this technology and increase the typical read length by several fold to obtain full-length mRNA information in our different model systems,“ says Carlos Alfonso-Gonzalez, the first author of the publication. In addition to Drosophila, the Hilgers Lab also included a human model of the nervous system into their study: cerebral organoids – “mini-brains” cultured in a dish from induced pluripotent stem cells.
Transcription end sites are pre-determined at transcription start
The gathered data representing each mRNA at the full-molecule scale give unprecedented insight into the transcription of individual genes “We realized that far from start sites (TSSs) and end sites (TESs) being randomly combined one to another, we found that often, sites of transcription start are specifically linked to distinct sites of transcription end”, says Valérie Hilgers. This linkage is actually causal: in ovaries, for example, the artificial activation of a TSS that is normally only used in the brain overrides the normal TES and artificially induced the use of the brain TES. This shows the critical role of TSS in shaping the RNA landscape unique to each tissue, and thereby influencing tissue identity.
Promoter dominance drives RNA diversity, gene function and tissue identity
However, one phenomenon stood out. “Certain TSSs show unexpected dominance behavior. They overrule conventional signals to end transcription, outcompete other TSSs, and cause the selection of distinct TESs. Accordingly, we named them »dominant promoters«,” says Carlos Alfonso-Gonzalez. Furthermore, the team found that interactions between these dominant promoters and their associated gene ends was guided by distinct epigenetic signatures. Importantly, the results in Drosophila brain cells could be replicated in the human brain organoids, showing that promoter dominance is a conserved, perhaps universal, mechanism for regulating the production of functional proteins and the cells’ functionality.
What could be the physiological relevance of this novel mechanism? Through an in-depth sequence conservation analysis, the Freiburg researchers discovered that TSSs and TESs exhibit co-evolution: over millions of years of evolution between species, individual nucleotide changes in the gene start at dominant promoters were accompanied by changes at the corresponding gene end. “We interpret this observation as a “push” through evolution, to sustain the interaction between both extremities of the gene, which implies significant importance of these couplings for animal fitness,” says Valérie Hilgers.
Journal
Cell
DOI
10.1016/j.cell.2023.04.012
Method of Research
Experimental study
Subject of Research
Animal tissue samples
Article Title
Sites of transcription initiation drive mRNA isoform selection.
Article Publication Date
12-May-2023



{kind=link}