Genome sequencing, where scientists use laboratory methods to determine a specific organism’s genetic makeup, is becoming a common practice in insect research. A greater understanding of insect biology helps scientists better manage insects, both those that are beneficial to the ecosystem and those that damage the food supply and threaten human health by carrying diseases.
Credit: Hidemasa Bono, Hiroshima University (CC-BY 4.0)
Genome sequencing, where scientists use laboratory methods to determine a specific organism’s genetic makeup, is becoming a common practice in insect research. A greater understanding of insect biology helps scientists better manage insects, both those that are beneficial to the ecosystem and those that damage the food supply and threaten human health by carrying diseases.
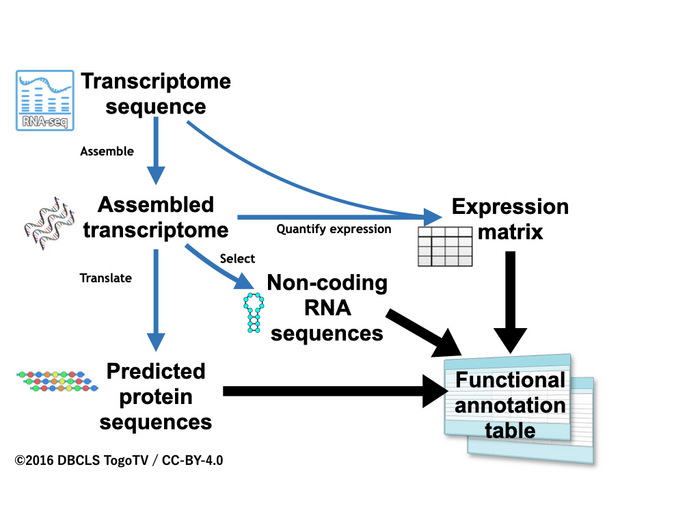
Researchers have developed a work-flow method, called Fanflow4Insects, that annotates gene functions in insects. In functional annotation, scientists collect information about a gene’s biological identity. The team’s new method uses transcribed sequence information as well as genome and protein sequence databases. With Fanflow4Insects, the team has annotated the functional information of the Japanese stick insect and the silkworm, including gene expression as well as sequence analysis. The functional annotation information that their workflow provides will greatly expand the possibilities of entomological research using genome editing.
The team, with scientists from Hiroshima University, Tokyo University of Agriculture and Technology, and RIKEN Center for Integrative Medical Sciences, has published their Fanflow4Insects method on June 27 in the journal Insects.
Insects are so diverse and abundant that scientists need a way of studying them on a large scale. This is what led scientists to begin work on sequencing the genome of insects. As of May 2022, scientists had decoded and registered the genomes of around 3000 insect species. They are also using long-read sequencing technology to further accelerate the pace of insect genome sequencing.
Next-generation sequencing has made it easier for researchers to decode the genomes of numerous insects along with their transcript sequences. However, the biological interpretation of these sequences remains a primary bottleneck of transcriptome analysis. The transcriptome is the sum of an organism’s RNA molecules. Transcriptome analysis is an important first step in functional annotation, which serves as an important clue for selecting genome editing targets.
Because some insects have genomes larger than the human genome, the difficult process of whole-genome sequencing is even more complicated. So scientists are using transcriptome sequencing with next-generation sequencing technology, also called RNA sequencing, as a tool for evaluating large genome-size insects. With this powerful tool, scientists can efficiently identify tens of thousands of possible genes in a specific tissue by assembling tens of millions of reads. They then assemble the gene sequences into transcriptional units for identification. But this type of analyses is dependent upon the scientists having access to comprehensive datasets and their functional annotation. Databases do exist, but they are unable to keep pace with the increase in insect genome sequencing.
As transcriptome analysis becomes more popular, many research groups are running their own pipelines, with the information regarding the transcription units from various studies being reported on a study-by-study basis. These pipelines are sets of algorithms used to processes the genome sequencing data. But scientists need a way to integrate the functional annotation from all the different groups doing this type of research into public databases.
In this current study, the research team used their newly developed Fanflow4Insects to create a functional annotation pipeline for the silkworm. Then the researchers also tested Fanflow4Insects for the transcriptomes of the Japanese stick insect. “Functional annotation is one of the most important processes to accelerate the selection of target genes once genome or transcriptome of the target organism is decoded. The functional annotation information obtained by the workflow Fanflow4Insects will greatly expand the possibilities of entomological research using genome editing,” said Hidemasa Bono, a professor with the Graduate School of Integrated Sciences for Life at Hiroshima University, and the first and corresponding author on the paper.
The Fanflow4Insects workflow for insects has been openly developed on GitHub, and is freely accessible. In conjunction with the functional annotation derived from expression, the data from Fanflow4Insects can be applied to the comparative study of insects with distinct phenotypes. “Using Fanflow4Insects, we are going to annotate insects that produce useful substances. The ultimate goal of this study is to make it possible to design molecular networks in insects using computer simulation,” said Bono.
###
The research team includes Hidemasa Bono, Hiroshima University; Takuma Sakamoto and Hiroko Tabunoki, Tokyo University of Agriculture and Technology; and Takeya Kasukawa, RIKEN Center for Integrative Medical Sciences.
This research was funded by a Japan Society for the Promotion of Science Kakenhi grant, an open innovation platform for industry–academia co-creation (COI-NEXT), Japan Science and Technology Agency, and the ROIS-DS-JOINT.
About Hiroshima University
Since its foundation in 1949, Hiroshima University has striven to become one of the most prominent and comprehensive universities in Japan for the promotion and development of scholarship and education. Consisting of 12 schools for undergraduate level and 4 graduate schools, ranging from natural sciences to humanities and social sciences, the university has grown into one of the most distinguished comprehensive research universities in Japan.
English website: https://www.hiroshima-u.ac.jp/en
Journal
Insects
DOI
10.3390/insects13070586
Article Title
Systematic Functional Annotation Workflow for Insects
Article Publication Date
27-Jun-2022

{kind=link}