Credit: Wilfried Guiblet and Dani Zemba, Penn State
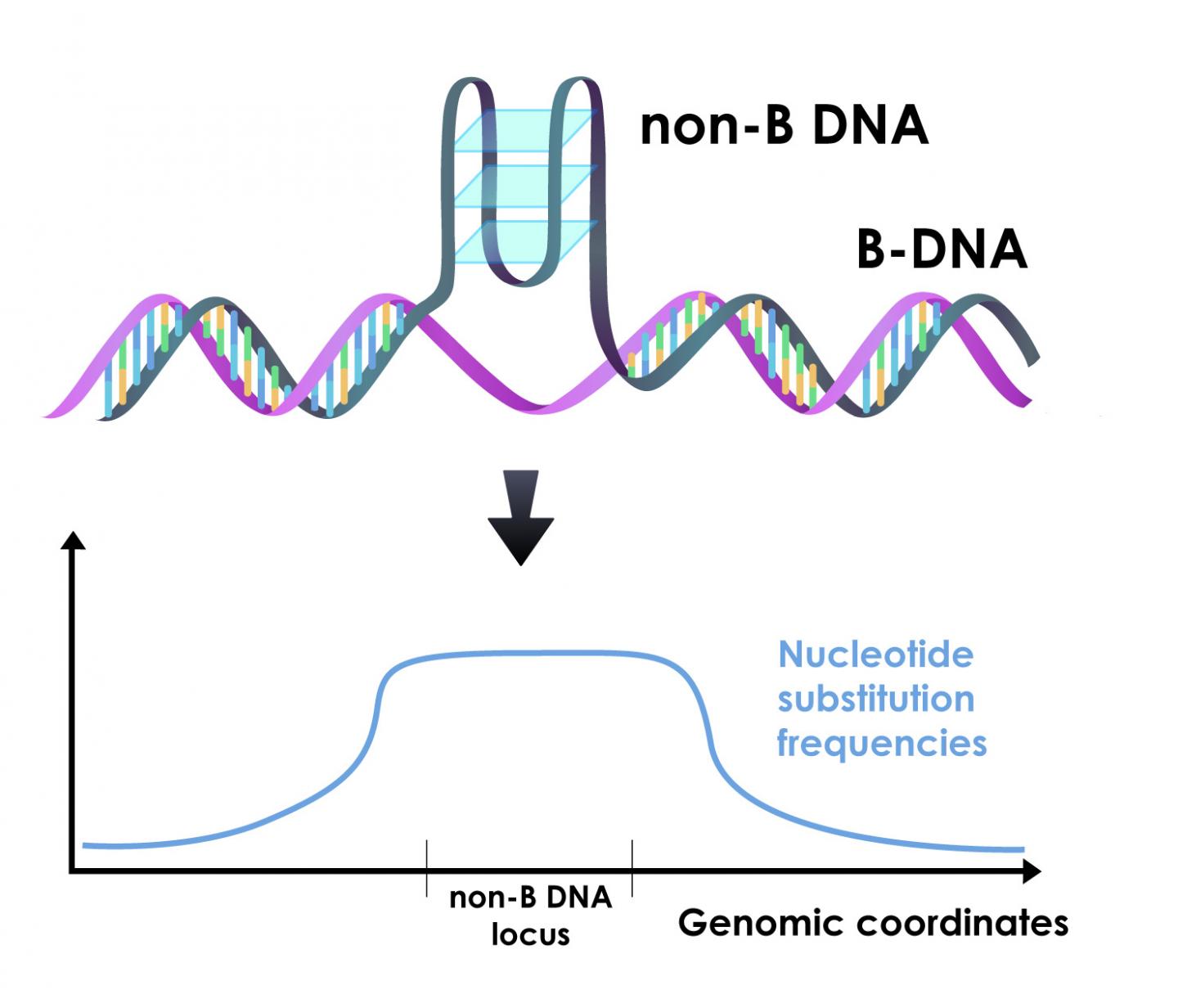
DNA sequences that can fold into shapes other than the classic double helix tend to have higher mutation rates than other regions in the human genome. New research shows that the elevated mutation rate in these sequences plays a major role in determining regional variation in mutation rates across the genome. Deciphering the patterns and causes of regional variation in mutation rates is important both for understanding evolution and for predicting sites of new mutations that could lead to disease.
A paper describing the research by a team of Penn State scientists is available online in the journal Nucleic Acids Research.
“Most of the time we think about DNA as the classic double helix; this basic form is referred to as ‘B-DNA,'” said Wilfried Guiblet, co-first author of the paper, a graduate student at Penn State at the time of research and now a postdoctoral scholar at the National Cancer Institute. “But, as much as 13% of the human genome can fold into different conformations called ‘non-B DNA.’ We wanted to explore what role, if any, this non-B DNA played in variation that we see in mutation rates among different regions of the genome.”
Non-B DNA can fold into a number of different conformations depending on the underlying DNA sequence. Examples include G-quadruplexes, Z-DNA, H-DNA, slipped strands, and various other conformations. Recent research has revealed that non-B DNA plays critical roles in cellular processes, including the replication of the genome and the transcription of DNA into RNA, and that mutations in non-B sequences are associated with genetic diseases.
“In a previous study, we showed that in the artificial system of a DNA sequencing instrument, which uses similar DNA copying processes as in the cell, error rates were higher in non-B DNA during polymerization,” said Kateryna Makova, Verne M. Willaman Chair of Life Sciences at Penn State and one of the leaders of the research team. “We think that this is because the enzyme that copies DNA during sequencing has a harder time reading through non-B DNA. Here we wanted to see if a similar phenomenon exists in living cells.”
The team compared mutation rates between B- and non-B DNA at two different timescales. To look at relatively recent changes, they used an existing database of human DNA sequences to identify individual nucleotides–letters in the DNA alphabet–that varied among humans. These ‘single nucleotide polymorphisms’ (SNPs) represent places in the human genome where at some point in the past a mutation occurred in at least one individual. To look at more ancient changes, the team also compared the human genome sequence to the genome of the orangutan.
They also investigated multiple spatial scales along the human genome, to test whether non-B DNA influenced mutation rates at nucleotides adjacent to it and further away.
“To identify differences in mutation rates between B- and non-B DNA we used statistical tools from ‘functional data analysis’ in which we compare the data as curves rather than looking at individual data points,” said Marzia A. Cremona, co-first author of the paper, a postdoctoral researcher at Penn State at the time of the research and now an assistant professor at Université Laval in Quebec, Canada. “These methods give us the statistical power to contrast mutation rates for the various types of non-B DNA against B-DNA controls.”
For most types of non-B DNA, the team found increased mutation rates. The differences were enough that non-B DNA mutation rates impacted regional variation in their immediate surroundings. These differences also helped explain a large portion of the variation that can be seen along the genome at the scale of millions of nucleotides.
“When we look at all the known factors that influence regional variation in mutation rates across the genome, non-B DNA is the largest contributor,” said Francesca Chiaromonte, Huck Chair in Statistics for the Life Sciences at Penn State and one of the leaders of the research team. “We’ve been studying regional variation in mutation rates for a long time from a lot of different angles. The fact that non-B DNA is such a major contributor to this variation is an important discovery.”
“Our results have critical medical implications,” said Kristin Eckert, professor of pathology and biochemistry and molecular biology at Penn State College of Medicine, Penn State Cancer Institute Researcher, an author on the paper, and the team’s long-time collaborator. “For example, human geneticists should consider the potential of a locus to form non-B DNA when evaluating candidate genetic variants for human genetic diseases. Our current and future research is focused on unraveling the mechanistic basis behind the elevated mutation rates at non-B DNA.”
The results also have evolutionary implications.
“We know that natural selection can impact variation in the genome, so for this study we only looked at regions of the genome that we think are not under the influence of selection,” said Yi-Fei Huang, assistant professor of biology at Penn State and one of the leaders of the research team. “This allows us to establish a baseline mutation rate for each type of non-B DNA that in the future we could potentially use to help identify signatures of natural selection in these sequences.”
Because of their increased mutation rates, non-B DNA sequences could be an important source of genetic variation, which is the ultimate source of evolutionary change.
“Mutations are usually thought to be so rare, that when we see the same mutation in different individuals, the assumption is that those individuals shared an ancestor who passed the mutation to them both,” said Makova, a Penn State Cancer Institute researcher. “But it’s possible that the mutation rate is so high in some of these non-B DNA regions that the same mutation could occur independently in several different individuals. If this is true, it would change how we think about evolution.”
###
In addition to Guiblet, Makova, Cremona, Eckert, Huang, and Chiaromonte, the research team at Penn State includes Robert S. Harris and Di Chen. The research was funded by the U.S. National Institutes of Health, Penn State Clinical and Translational Sciences Institute, the Penn State Institute of Computational and Data Sciences, the Huck Institutes of the Life Sciences at Penn State, the Penn State Eberly College of Science, the Pennsylvania Department of Health, and the CBIOS Predoctoral Training Program.
Media Contact
Sam Sholtis
[email protected]
Original Source
https:/
Related Journal Article
http://dx.

{kind=link}